Document Type : Original Research Article
Authors
Department of Chemistry, Faculty of Physical Sciences, Ahmad Bello University, Zaria, P.M.B.1045 Kaduna State, Nigeria
Abstract
V600E-BRAF mutation has been detected in several malignant tumours. Developing potent V600E-BRAF inhibitors is considered an essential step in curing diverse cancer types. In this work, computational techniques such as the molecular docking simulation, ADMET evaluation, and density functional theory (DFT) calculations were applied to screen and identify the most potential V600E-BRAF inhibitors from a series of 39 previously synthesized novel pyrrolo[2,3-b]pyridine derivatives. Five top-ranked compounds (3, 18, 32, 33, and 35) with remarkable docking scores, compared to Vemurafenib (FDA-approved V600E-BRAF inhibitor), were selected. The formation of H-bonds and hydrophobic interactions with critical residues for V600E-BRAF as Vemurafenib confirmed the high stability of these complexes. Subsequently, the compounds were screened by analyzing their drug-likeness and ADMET properties. The compounds possess safety agents and effective combination therapy as pharmaceutical drugs. The highest occupied molecular (HOMO) orbital, lowest unoccupied molecular orbital (LUMO), and energy gap values were calculated using the DFT. The molecular electrostatic potential (MEP) was analyzed to illustrate the charge density distributions that could be associated with the biological activity. Therefore, compound 35 emerged as a potential hit with enhanced pharmacological properties and could be presented as a promising cancer drug candidate.
Graphical Abstract
![Exploration of Anticancer Potential of Novel Pyrrolo[2,3-b]pyridine Derivatives Targeting V600E-BRAF Kinase: Molecular Docking, Pharmacokinetic and DFT Studies](javascript:loadModal('Exploration of Anticancer Potential of Novel Pyrro ... ', 'data/ajcsa/coversheet/971662744258.jpg'))
Keywords
Introduction
Cancer is a type of disease that causes a threat to human health and human1life [1,2]. In recent years, the morbidity rate of cancer has increased [3,4]. Cell proliferation is triggered by activating the MAPK signaling pathway [5,6]. A cascade of protein kinases regulates the activation of the MAPK signaling pathway phosphorylated successively and controls several cellular activities such as; cell proliferation, differentiation, and migration [1,7]. BRAF is a kinase that plays an essential role in MAPK singling pathway. The most common mutation in BRAF is V600E-BRAF which was identified in 8% of all cancer types, such as; thyroid cancer (30–70%), melanoma (60%), and colorectal cancer (10%) [3,8]. Therefore, the BRAF kinase enzyme is considered a crucial biological target in treating and controlling cancer disease [4,9]. Vemurafenib and dabrafenib were approved by the U.S. Food and Drug Administration (FDA) to treat late-stage melanoma. Both are selective inhibitors of V600E-BRAF kinase and cause programmed cell death in melanoma cell lines, hence high potency against melanoma cell lines [10,11]. Mono-therapy treatment with V600E-BRAF significantly improved both survival rate and patient’s lifestyle.
Despite the success of selective V600E-BRAF inhibitors, this success is short-lived, and resistance to these inhibitors appears from 15 to 8.8 months [12,13]. The acquired resistance to selective V600E-BRAF makes the development of new candidates to be a new therapy for V600E-BRAF embedded cancers an essential issue. In drug discovery research, the identification and validation of lead compounds and the determination of active binding sites of biological targets related to a particular lead compound performed through wet lab experiments are pretty expensive and time-consuming [14]. Computational approaches effectively reduce the time required to obtain valuable drugs and decrease their associated economic costs, making it possible to propose new potential drugs with low expenditures and selective targeting [15]. The molecular docking method is widely used to determine the proper orientation of drug molecules in the protein-active site and to measure their binding affinity. Previously, extensive molecular docking studies were conducted to explore the biological activity of many chemical materials [16-18]. Drug likeness criteria and an in silico ADMET profile are two other virtual screening tools for adopting compounds that may exhibit physiological activity and drug-like identity. The protocols used to estimate drug-likeness and ADMET features are produced from a variety of experimental sources of data that have been documented in drug databases [19, 20].
Density functional theory (DFT) is the most popular method, with a lower computational cost than many other methods. DFT calculations currently yield the most precise and reliable results for different material systems, which are pretty compatible with the experimental data [21-23]. In the present research study, thirty-nine novel pyrrolo[2,3-b]pyridine derivatives recently added to the literature by Abdel-Maksoud et al. [24] were investigated computationally for their potential to inhibit the V600E-BRAF kinase from exploring their anticancer activity and adding more drug candidates to the medicinal chemistry library. The studied compounds exhibited high enzymatic and cellular activities upon testing against the V600E-BRAF kinase enzyme and NCI sixty human cancer cell lines. The study performed a systematic computational exploration of these compounds using molecular docking simulation, ADMET prediction, and DFT calculations. The main objective is to evaluate the anticancer capacity of these chemical structures as potential drug with potent pharmacological properties.
Materials and Methods
Ligand selection and preparation
Thirty-nine novel pyrrolo[2,3-b]pyridine derivatives were retrieved from the literature [24]. Structures of ligands were drawn using ChemDraw (Table S1), and energy minimization was done using MM2 force field in Spartan 14 to help the docking program detect the bioactive conformer from the local minima. The compounds were optimized using the DFT method at the B3LYP level of theory and 6–31G* (d, p) basis set, then saved in a PDB file format for the docking.
Receptor preparation and molecular docking simulation
The crystal structure of V600E-BRAF (PDB-ID: 3OG7) [25] and the native ligand (vemurafenib) was collected from the Protein Databank at (http://www.rcsb.org/). The PDB file of V600E-BRAF was prepared with Molegro Virtual Docker 6.0 [26] by updating the H-atoms and eliminating the water (excess) molecules found in the complex structure; Vemurafenib was also detached from the target. The potential ligand-binding cavity of the V600E-BRAF receptor was predicted, and the binding cavity was set inside a restricted sphere of X: 1.59, Y: - 1.28, Z: -6.2 with a radius 28 Å having a grid resolution of 0.30 Å. For1molecular docking, all the prepared compounds (ligands), including vemurafenib (reference-inhibitor), were then imported into the Molegro Virtual Docker 6. Discovery Studio (DS) Visualizer was adopted to visualize various intermolecular interactions.
Physico-chemical and ADMET biochemical investigation
Selected compounds from the molecular docking analysis were assessed for their drug-like behavior through an online server (SwissADME) [27], and the analysis of pharmacokinetic parameters required for absorption, distribution, metabolism, and excretion (ADME) was performed using the online server (pkCSM) [28].
The DFT calculations
The electronic and structural properties of the five best hit compounds selected from the docking investigation were calculated using the DFT method at the B3LYP level of theory and 6–31G* (d,p) basis set aided by Spartan 14 software. The calculated parameters used in this study include the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energies, hardness (η), softness (σ), electronegativity (χ), chemical potential (μ), and electrophilicity index(ω) [29]. The molecular electrostatic potential surfaces (MEPs) were obtained from the population analysis calculations and visualized using Spartan 14 software. These parameters play a significant role in elucidating the magnitude of ligands interaction in the binding pocket of V600E-BRAF receptor.
Result and Discussions
To screen and identify potential V600E-BRAF kinase inhibitors, native ligand (vemurafenib) and all the studied 39 ligands were docked into the binding pocket of the target, in which docking scores were served in terms of MolDock and Rerank scores. Moreover, the binding score of the native ligand was selected as a benchmark. From the molecular docking simulation studies of novel pyrrolo[2,3-b]pyridine derivatives against V600E-BRAF (PDB ID: 3OG7), it is revealed that most of the studied ligands docked at the active site of the enzyme with a favorable Rerank score and docking score compared to vemurafenib (Table S2). Five of these ligands exhibited superior binding scores and good interaction compared to vemurafenib, as demonstrated in Table 1. Identifying key contributing residues in the binding pocket of the V600E-BRAF kinase of the five selected complexes (3,18,32,33 and 35) was performed using the Discovery Studio Visualizer. The residues involved in the molecular interactions of the five best-docked ligands with V600E-BRAF kinase are presented in Table 2. The strength of the ligand-protein interaction is considered based on the Rerank score, which is defined as a linear combination of E-inter, which is of Van der Waals, steric, electrostatic, hydrogen bonding, the energy between the ligand and the protein, and E-intra, which is of Van der Waals, hydrogen bonding, torsion, electrostatic, sp2–sp2 energy of the ligand weighted by pre-defined coefficients [26, 30].
Table 1. Docking results of the five best-docked ligands in V600E-BRAF kinase (PDB ID: 3OG7)

From the docking result, the selected ligands formed bonds and non-bond interaction at the binding cavity of the V600E-BRAF, as evident from the interaction energy and hydrogen bonding energy (Table 1). The docked results showed that inhibitors swing between the space of hydrophobic residues; PHE468, PHE583, TRP531, LYS483, ASP594, ILE463, ALA481, LEU514, CYS532, VAL471, and LEU514, which gave more conformational freedom, on the one hand; residues SER536, ASP594, GLY534, TRP531, CYS532, LYS48, CYS532, ALA481, ILE527, LEU514, and THR529 showed hydrogen bond interaction with the ligands as shown in Table 2. Also, protein ligands interaction showed that amino groups, sulphonamides terminal, imidazole, pyridine, and benzyl rings played an important role. The details of the complex structures with the five best docking scores are provided in Figures 1 and 2.
The benzene ring of ligand 3 in complex 3 (MolDock score: -139.129 kcal.mol-1 and Rerank score: -115.982 kcal.mol-1 as shown in Table 1) formed π-alkyl interaction was formed with PHE595, LEU505, LEU514, VAL471, ALA481, LEU514, ILE463, VAL471, and ILE463 residues. Meanwhile, one of the N atom donors of the sulphonamide moiety formed a1conventional hydrogen bond with LYS483. The O atom of the sulphonamide conserved residue ASP594, with a bond length (b.l) of 2.53232 Å. Carbon hydrogen bonds are formed between the pyridine ring and NH group with SER536 and GLN530, as presented in Table 2. Apart from H-bonding, the pyrrole ring escalated to form hydrophobic π-π interaction with TRP531 and PHE583. Others include π-Sulphur interaction with CYS532. These hydrophobic and H-bonding interactions of V600E-BRAF with the ligand may account for the good binding affinity (Figure 1, complex 3).
Ligand 18 inhibits V600E-BRAF with a MolDock and Rerank score of -143.937 kcal.mol-1 and -119.938 kcal.mol-1, while the E-Hbond was -4.682 kcal.mol-1 (Table 1). The carbonyl O atom bonded to –NH group exhibit three H-bonds with GLY596, PHE595, and ASP594 (b.l 1.71168, 2.87511, and 2.5981 Å, respectively). Meanwhile, PHE583 and TRP531 were sandwiched between the pyridine through π-π interactions (Figure 1, complex 18), which also contributes to the stabilization of the complex. LYS483 formed π-cation interaction with the ligand. Moreover, CYS532 interacted with ligand 18 through π-sulfur hydrophobic interaction (Table 2).
Table 2. Molecular interactions of the five best docked ligands with V600E-BRAF kinase


 Figure 1. 3D and 2D representations for the interactions of selected complexes (3, 18, and 32)
Figure 1. 3D and 2D representations for the interactions of selected complexes (3, 18, and 32)
In the docked complex of ligand 32 (MolDock score: -130.690 kcal.mol-1, Rerank score: -104.308 kcal.mol-1), as presented in Table 1, the O atom of the nitro group formed a conventional H-bond with N atom of LYS483 (b.l 2.3228 Å), the N atom of the pyrrole ring interact with the GLN530 via H-bonding (b.l 1.98675 Å). Meanwhile, the N of the pyridine ring exhibited an H-bond network with CYS5321 (b.l 2.39133 Å). The Br atom bonded to the pyridine ring formed π-alkyl interaction with the side chain of TRP531 and ILE463; the nitro group attached to the benzene ring formed a Carbon-H-bonding interaction with ASP594. Pyridine ring formed π-π interaction with the side chain of the TRP531 similar to vemurafenib, as shown in Table 2. From the above results, it can be pointed out that H-bond interactions with the key binding residues are significant motivators for stabilizing the inhibitor within the catalytic pocket (Figure 1, complex 32).
A MolDock score of -149.304 kcal.mol-1 and Rerank score of -116.838 kcal.mol-1 were obtained for the docking of ligand 33 as in Table 1 in the binding site of V600E-BRAF (Figure 2, complex 33) indicates strong interactions between the ligand and the receptor. The O atom that linked the two –N atoms of the benzene ring interacts with GLY596 and ASP594 via H-bonds (b.l 1. 2.40015 and 2.56379 Å). Also, the –NH group of the pyrrole ring and F atom interact with ASN581 and ASP594 all through the conventional H-bond. The pyridine ring and the F atom attached to the benzene ring extend to form C-Hbond with the receptors (ASP594 and GLY596). The pyrrole ring stacks vertically, forming a π-π interaction with PHE583. This complex has additional stability by forming π-alkyl interactions with residues LEU514, LYS483, and VAL471. The appearance of other interactions such as π-cation, and π-sigma, as demonstrated in Table 2, and complex 33 also assists with strong binding inside the active site of V600E-BRAF.
 Figure 2. 3D and 2D representations for the interactions of selected complexes (33, 35 and Vem.).
Figure 2. 3D and 2D representations for the interactions of selected complexes (33, 35 and Vem.).
The docked complex of ligand 35 in the binding pocket of V600E-BRAF revealed an excellent docking score (MolDock score: -179.617 kcal.mol-1 and Rerank score: -136.819 kcal.mol-1) as presented in Table 1. The –N atom of the pyrrole and pyridine ring formed a conventional H-bond with GLN530 and CYS532 (b.l 2.43723 and 2.93751 Å), the carbonyl oxygen adjacent to –N atom also interacted with LYS483 via H-bonding (b.l 1.84074 Å). A halogen bond is formed with the –F atoms of the ligand. The pyridine ring, pyrrole ring, and benzene ring formed π-alkyl interaction with the side chain of LEU505, LEU514, LYS483, ALA481, LEU514, CYS532, ALA481, and CYS532; the pyrrole ring also formed Carbon-H-bonding interaction with the side chain of CYS532 and THR529 as shown in Table 2. The above results indicate that H-bond interactions with the key binding residues are significant motivators for stabilizing the inhibitor within the binding pocket (Figure 2, complex 35).
H-bonding is a significant indicator of protein-ligand solid interactions and commonly results in high binding affinity [31]. In protein-ligand interactions, the number of hydrogen bonds often increases the inhibitor potency against the target protein. Figures 1 and 2 indicate a 3D and 2D representation of the binding modes within the target protein’s active site. The formation of conventional hydrogen bonds of the selected ligands with anticancer target protein resulted in good ligand binding. Furthermore, as a standard comparison with the studied ligands, the melanoma drug (Vemurafenib), which also contains sulphonamide moiety, imidazole, and pyridine ring, was docked into the same investigated protein receptor and specifically compound 35 among the selected ligands outperforms Vemurafenib. Remarkably, it inhibited the melanoma target more effectively than Vemurafenib. The selected ligands had a higher docking score, a more stable MolDock score, and a much lower Re-rank score than Vemurafenib.
Drug-likeness analyses and ADME predictions for investigated ligands were performed via SwissADME and pkCSM web servers [27, 28]. The results for the predicted drug-likeness properties are presented in Table 3, and that of ADMET properties are given in Table 4. Drug-likeness analyses and ADME predictions were also performed for the reference drug (Vemurafenib) to compare. According to the Lipinski rule, good absorption or permeation is more likely when: the molecular weight (Mol.Wt.) <500, hydrogen bond donors (HBDs) <5 (counting the sum of all NH and OH groups), partition coefficient octanol/water Log P < 5, the number of hydrogen bond acceptors (HBAs) <10 (counting all N and O atoms). Results presented in Table 3 showed that the molecular weights of the investigated compounds are in the range of 376.39 to 428.90 g.mol-1. Thus, the compounds obeyed the Lipinski rule. It also showed that none of the investigated compounds have more than ten hydrogen bond acceptors. The highest number of hydrogen bond acceptors was obtained for compounds 3 and 35 with the value of 6, as illustrated in Table 3. On the other hand, it was observed that all the selected compounds have less than five hydrogen bond donors, and the Log P value was predicted to be < 5 (Table 3).
According to Ghose rule, the partition coefficient for a given molecule should be between -0.4 and 5.6. In the study, the average partition coefficient of the molecules, which is the average of Log P were predicted to be in the range of 2.93-3.52, as shown in Table 3. According to the Veber rule, topological polar surface area should not be greater than 140 Å2. The study observed that the investigated compounds' polar surface areas are not greater than 110.18 Å2 (Table 3). According to Veber and Muegge's rules, rotatable bonds in the molecule should not be more than 10 and 15, respectively. All the molecules investigated in the study have 3 to 7 rotatable bonds, as shown in Table 3, and obey the Veber and Muegge rules [32]. The selected compounds were further screened for an optimum profile of permeability and bioavailability using a bioavailability score (ABS) criteria. An ABS score of 0.55 indicates obedience to the Lipinski rule [33].
Table 3. Predicted Drug-likeness properties of the selected ligands

Table 4. Predicted ADMET properties of the selected ligands.

The studied ligands have an intestinal absorbance value in the range of 86.900 to 96.994%, as presented in Table 4, indicating their ease of absorption. VDss stands for volume of distribution at steady state, which is the apparent volume of distribution after enough time has passed for the drug to distribute uniformLy through all tissues. A high VDss value (>0.5) indicates that the drug is well distributed in the plasma, whereas a low VDss value (<-0.5) indicates that the drug has a poor ability to cross the cell membrane. The predicted VDss value in Table 4 ranges from -0.232 to 0.305, indicating that the studied ligands have a reasonable plasma distribution. Also, the blood-brain barrier (BBB) and the permeability of the central nervous system (CNS) are important factors in achieving the optimum pharmacological drug. Based on the standard ranking of drug the BBB and CNS permeability standard values are given as > 0.3 to < -1 log BB and > -2 to < -3 log PS. For a given ligand, log BB < - shows poor drug distribution to the brain, while a value of log BB > 0.3 implies that the drug can cross the BBB and log PS, and a value > -2 implies that the drug candidate can penetrate the CNS. In contrast, a value < -3 indicates that it will be difficult for the drug candidate to induce into the CNS [34]. The results in Table 4, indicated that the selected compounds had shown a high potential to cross barriers.
Cytochrome P450 is a vital metabolizing enzyme in the human body with five major isoforms: CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4. The results in Table 4 reveal positive inhibition capability for these enzymes; therefore, it is safe in pharmacokinetic interactions. The bioavailability of a drug and its dosing rates to reach steady-state concentrations is measured by total clearance. The faster the molecule’s excretion, the higher the total clearance value. AMES toxicity is used to determine whether a molecule is mutagenic or not. The results in Table 4 indicate that compounds 33 and 35 are nontoxic. Thus, it can be concluded that the selected ligands possess the desired pharmacokinetic properties and can be used as V600E-BRAF inhibitors and drugs against melanoma cancer in the future.
Optimized geometries of the selected ligands obtained from DFT calculations are given in Figure 3. Results showed that all the geometry-optimized structures correspond to a global minimum. Frontier molecular orbitals (HOMO and LUMO diagrams) of the five-hit ligands specify a crucial role in charge-transfer interactions with the binding site of V600E-BRAF. As presented in Figure 4, the blue and red colors show the positive and negative values of the orbital. More so, the shapes of the frontier orbitals can be used as a guide in determining reactivity. On every ligand, the HOMO is delocalized onto the imidazole and two adjacent benzyl rings and mainly dominated on the pi-bonds. By convention, the blue region represents the maximum value of the HOMO, and the red region represents the minimum value. The HOMO electron density distribution of the studied ligands indicates favorable interactions of the ligands to the V600E-BRAF. Likewise, the LUMO delocalizes over several sites of pyridine and imidazole rings. Still, the enormous contribution comes from one N-atom and the conjugated bonds of the pyridine ring for ligands 3 and 32. While for the other ligands (18, 33, and 35), the LUMO was dominantly spread on the benzyl ring attached to the sulphonamide terminal. The electronic surfaces of HOMO and LUMO revealed that the pyridine and imidazole rings can interact with the target under favorable conditions. This typical behavior is suitable for donor-acceptor interactions, which may be responsible for the excellent binding of the ligands to the V600E-BRAF.
 Figure 3. Optimized geometric structures of the studied ligands (3, 18, 32, 33, and 35)
Figure 3. Optimized geometric structures of the studied ligands (3, 18, 32, 33, and 35)

Figure 4. Frontier molecular orbital surfaces of the studied ligands (3, 18, 32, 33, and 35)
Table 5 shows the HOMO and LUMO energies and the associated Quantum chemical descriptors of the studied ligands. The higher HOMO value denotes a molecule with a good electron-donor, whereas a lower value implies a weak electron-acceptor. Furthermore, a smaller energy gap between the LUMO and HOMO energies considerably influences molecules' intermolecular charge transfer and bioactivity. Thus, a small energy gap observed in the hit ligands positively affects the electron moving from the HOMO to the LUMO, leading to a strong affinity of the inhibitor for V600E-BRAF. The Egap value increases according to the following: 18 (3.15 eV) > 32 (4.05 eV) > 33 (4.32 eV) > 35 (4.41 V) > 3 (4.70 eV). Hence, the reactivity order increases accordingly, where the most reactive is 18 (3.15 eV). The order of reactivity increases conforms to the decreases in energy gap values.
Table 5. Frontier molecular orbital energies and global reactivity parameters of the selected ligands

The hardness (η) and softness (σ) are essential descriptors for the behavior of a molecule in a chemical reaction., Complex molecules have a high resistance to changing their electronic distribution during a reaction. In contrast, soft molecules have a low resistance to changing their electronic distribution during a reaction. The obtained results revealed a high hardness value plus a low softness value compared to similar structures found in the literature [35]. The electronegativity (χ) of a molecule measures its ability for electron attraction [36]. The electronegativity was calculated to be in the range of 3.19 to 3.76 eV, describing the studied ligands as donor-electrons. The chemical potential (μ) indicates negative values for all the studied ligands, which implies good stability, and the formation of a stable complex with the receptor. Electrophilicity (ω) is a predictor of the electrophilic nature of a chemical species; it measures the propensity of a molecule to accept an electron, with high values of ω characterizing good electrophilicity in a molecule. The following is a ranking of organic molecules according to electrophilicity; weak electrophiles have ω less than 0.8 eV, moderate electrophiles have ω in a range between 0.8 and 1.5 eV, and strong electrophiles have ω greater than 1.5 eV [37]. As a result, the calculated electrophilicity value characterizes the investigated ligands as good electrophiles. It is common knowledge that highly electrophilic drugs have potent anticancer properties [38]. Finally, comparing the values of molecular orbital energies (eV), global reactivity descriptors, and docking scores of five-hit ligands may be considered potential V600E-BRAF inhibitors.
The molecular electrostatic potential (MEP) surface describes the charge distribution, which gives good insight into the physical and chemical properties of the molecule. It also predicts reactive sites for electrophilic and nucleophilic attacks in a molecule. If the point charge is located in a region of excess positive charge, the point charge-molecule interaction becomes repulsive, and the electrostatic potential will be positive. However, if the point charge is located in an excess negative charge region, the interaction is attractive, and the electrostatic potential becomes negative. The MEP maps of the investigated ligands obtained from DFT calculations are given in Figure 5. From the map, red indicates the nucleophilic region, blue indicates the electrophilic region, and the colors between indicate intermediate values of the MEP. Therefore, the potential increases in the order: red<orange<yellow<green[39].
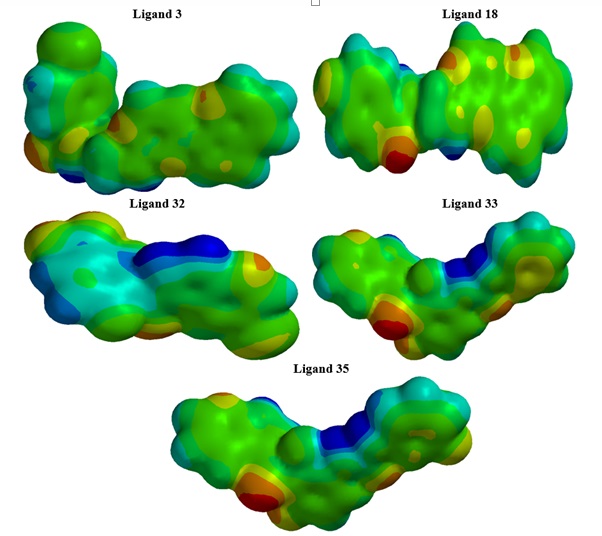
Figure 5. Molecular electrostatic potential (MEP) of the studied ligands (3, 18, 32, 33, and 35)
Conclusions
In this research study, molecular docking simulation was successfully used to screen and identify potential hits against V600E-BRAF from a series of novel pyrrolo[2,3-b]pyridine derivatives. Five top-ranked compounds (3, 18, 32, 33, and 35) were selected by docking studies, and their docking scores were superior to the native ligand (Vemurafenib) in the active pocket of the V600E-BRAF kinase. The docking results showed that H-bonds and hydrophobic interactions might play essential roles in the molecular interactions between the active compounds and the V600E-BRAF kinase. The predicted physiochemical and ADMET parameters of all the selected compounds were within the acceptable optimal requirements for drug development. Using the DFT approach, the quantum chemical descriptors recognized the selected molecules as stable structures, and MEP distribution revealed the potential sites for electrophilic/nucleophilic attacks. This research revealed that compound 35 may be considered a potential hit as an anticancer agent and can be selected for further studies like modification of the scaffold, characterization, and in vitro evaluation. These findings have agreed with the experimental reports as the selected hit exhibited cytotoxic activity against a different panel of human cancer cell lines.
Funding
The Tertiary Education Trust Fund fully sponsored this work (TETFUND (TETF/DR&D/UNI/ZARIA/IBR/2020/VOL. /54).
Acknowledgments
The authors thank the Tertiary Education Trust Fund (TETFUND) for the 2020 Institution Based Research (IBR) Project Grant (TETF/DR&D/UNI/ZARIA/IBR/2020/VOL.1/54) awarded for this research. The authors acknowledge Ahmadu Bello University, Zaria-Nigeria, for the technical support.
Disclosure statement
The authors reported no potential conflict of interest.
Orcid
Abdullahi B. Umar : 0000-0003-0984-5969
Adamu Uzairu : 0000-0002-6973-6361
)