Document Type : Original Research Article
Authors
1 Department of Pure and Applied Chemistry, Faculty of Science, University of Maiduguri, P.M.B. 1069, Maiduguri, Borno State, Nigeria
2 Department of Chemistry, Faculty of Physical Sciences, Ahmadu Bello University, P.M.B.1044, Zaria, Kaduna State, Nigeria
Abstract
The appearance of severe acute respiratory syndrome coronavirus 2 (COVID-19) is at its peak; with the growing number of people infected with COVID-19, there is an urgent need to find effective treatments for this outbreak. The current situation appears to call for drug repurposing. In our hunt for a viable medication against this virus, we used an in-silico strategy to test four conventional medicines, including Ritonavir and Hydroxychloroquine, against the spike glycoprotein of COVID-19. A docking simulation was performed to assess the drug's binding affinity. We discovered a single medication compound 36 against SARS-coronavirus spike glycoprotein. The compound was found to have a strong binding affinity against the target protein. The chemical was discovered to have a high affinity for the target protein. Furthermore, no conventional medicines efficiently bonded to the SARS-coronavirus spike glycoprotein. The current investigation concluded that the compound 36 is a highly stable anti-SARS-coronavirus spike glycoprotein medication. Furthermore, none of the standard drugs had a high affinity for the SARS-coronavirus spike glycoprotein binding site.
Graphical Abstract
![(S)-N-(4-Carbamoylphenyl)-8-Cyclopropyl-7-(Naphthalen-1-ylmethyl)-5-Oxo-2,3-Dihydro-5H-Thiazolo[3,2-a]Pyridine-3-Carboxamide, A Better Inhibitor of SARS-Cov-2 Spike Glycoprotein Than Some Standard Drugs: A Computational Prediction](javascript:loadModal('(S)-N-(4-Carbamoylphenyl)-8-Cyclopropyl-7-(Naphtha ... ', 'data/ajcsa/coversheet/21668447927.jpg'))
Keywords
Introduction
In 2019, the novel SARS-coronavirus emerged in Wuhan City, Hubei Province, China, hurting millions of people socially, economically, and mentally [1, 2]. As of August 12, 2022, World Health Organization (WHO) had received reports of 585,950,085 confirmed cases of COVID-19, including 6,425,422 deaths (https://covid19.who.int/more-resources) [3]. SARS-coronavirus is transmitted from person to person by respiratory droplets, direct human-to-human contact, high aerosol concentrations, and feces or urine on rare occasions [1, 4, 5]. The SARS-Cov-2 epidemic has affected the world for over three (3) years, with disastrous hygienic, economical, and sociological effects [6]. The current SARS-Cov-2 (Covid-19) crisis has proved that globally we are not prepared to respond with remedies to treat existing infections as there is no food and drug administration (FDA) approved therapeutic [7]. The current SARS-Cov-2 outbreak is already proving remarkably costly in terms of mortality and financial ramifications. It has recently been proposed that SARS-Cov-2 could cause the resurrection of other latent viruses, including HIV, STI, common cold, and HHV-6, which is liable for various skin symptoms primarily assigned to COVID-19, such as rashes and pityriasis rosea-like eruptions [8, 9]. The SARS-coronavirus can infect the lungs, kidneys, heart, and gut cells, causing organ damage and numerous organ dysfunction syndromes [1].
Biochemical and genetic studies have shown that the enzyme spike glycoprotein is essential for SARS-coronavirus survival [10, 11]. It has been revealed that the spike glycoprotein of SARS-Cov-2 possesses a strong affinity for binding to human ACE-2 receptors [12], and it was found to have a potential role in a viral entry inside the host [10]. Preventing SARS-coronavirus entrance into host cells via spike glycoprotein receptors could be an excellent way to combat SARS-Cov-2 [13].
In this context, spike glycoprotein has been recognized as a promising target for the current investigation. Hanson et al. [14] performed medication repurposing on 3384 small-molecule pharmaceuticals with 25 hits using a proximity-based test that assesses the binding of SARS-Cov-2 spike glycoprotein. In their unbound states, the S protein and ACE-2 do not contain therapeutic target pockets; however, they have well-defined pockets in their bound forms that can be used for drug development. Patil et al. [15] used computational methods to show that some antiviral medications used to treat hepatitis C (HCV) and human immunodeficiency virus (HIV) could function as immediate investigational molecules and possibly as potential candidate inhibitors. A computational technology called virtual screening aims to hasten the discovery of novel medicines.
This computational technology enables the scientific exploration of potential medication candidates by evaluating and directing choices of chemical structures with modified biological applications [16, 17]. The excellent synthon of thiazolino-2-pyridone amide derivatives enables the synthesis of various novel compounds with potential pharmacological characteristics [18]. Due to a wide variety of structural modifications (S)-N-(4-carbamoylphenyl)-8-cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-2,3-dihydro-5H-thiazolo[3,2-a]pyridine-3-carboxamide one of the derivatives of thiazolino-2-pyridone amide, has shown promising therapeutic efficacy and has been applied to cure sexually transmitted infections such as Chlamydia trachomatis infectivity [19, 20]. As such, the compound would be used for this current research. Molecular docking makes use of computer techniques to quantify ligand-protein free energy. Several spatial conformations of the complex are acquired, allowing evaluation of the most stable conformation (lowest energy) and the most significant inhibitory effect [21]. Virtual screening was used in this study to look for possible inhibitors of the SARS-Cov-2 spike (S) glycoprotein, which is responsible for attaching to the host cell receptor and causing membrane fusion [22]. Furthermore, the simulated screening compounds' pharmacokinetic and toxicological properties were studied.
Computational Details
This research chose a group of compounds for docking simulation and ADME predictions. The structures of five drugs (Table S1) that are currently understudies related to the inhibition of SARS-coronavirus SP were selected from our previous publications [19, 20, and 23] as a foundation for molecular docking simulation. The structures were created in two dimensions using the free program MarvinView Europium.6 [24], and Gaiuiasn 09 software was used to optimize [25]. The density functional theory (DFT) with Becke three-parameter hybrid functional combined with Lee-Yang-Parr correlation functional (B3LYP) and the 6-31G basis set approach to find the optimal structural conformation was used.
Homology modeling is a highly effective technique when interesting protein crystal structures are unavailable. It is the most valuable research tool for filling the sequence structure gap in structure-based drug design [26]. The homology modeling of the SARS-coronavirus-2 was built using crystal structures of spike glycoprotein (accession ID: QIG55955.1) which were retrieved from the National Center for Biotechnology Information (NCBI). Then the BLAST protein (http://blast.ncbi.nlm.nih.gov) program was used to search for a suitable template in Brookhaven Protein Data Bank (PDB) format. The PDB entries shown in (figure 1) were selected as templates for spike glycoprotein (SARS-coronavirus-2). The query sequence (accession ID: QIG55955.1) and template sequences were aligned using the MUSCLE [27, 28] method employed using the MEGA X program [29]. Phylogenetic analysis is utilized to determine the evolutionary links among the protein sequences. The phylogenetic analysis and subregion trees were performed using the UPGMA method [30] implemented in MEGA-X [31]. MODELLER v10.3 [32] was used to build homology models of spike glycoprotein SARS-Cov-2 crystallographic structure. Using the techniques included in MODELLER, 3D models of all non-hydrogen atoms were generated automatically from the alignments. The model with the lowest modeler objective (low discrete optimized protein energy score) and the fewest constraint violations was chosen for further examination from the five models produced by MODELLER for each alignment. The overall stereochemical quality of the final developed model for SARS-Cov-2 spike glycoprotein and the best template structure was assessed by the program ERRAT [33] and PROCHECK [34, 35]. The environment profile of the final developed model and the template were checked using Verify-3D [36] and ProSA [37]. The modeled and the template protein structures were undertaken for energy minimization by utilizing Swiss-PdbViewer (http://www.expasy.org/spdbv/). The energy-minimized proteins were moved to AutoDockTools v1.5.7 where the proteins were saved in PDBQT file format. The grid was fixed around the active site, and score prediction was obtained from the compute pocket [AutoSite 1.1] implemented in AutoDockFR v1.2 [38]. Molecular docking simulation studies were carried out in AutoDock-Vina [39] and implemented in EasyDockVina v2.2. The docking was performed within a restricted search space with a center and dimension set, as displayed in Table 1.
In Silico Pharmacokinetics ADMET Prediction
Pharmacokinetics is the study of how a medication behaves within the body. It is feasible to select the routes of administration and modify the dosages for a drug's future use by knowing the pharmacokinetic properties of that substance [40-42]. Absorption, distribution, metabolism, and excretion are the four essential activities that determine a compound's in vivo pharmacokinetics (ADME) [43]. Following a docking studies analysis to investigate the binding affinity and kind of contact, ligands that showed a strong binding affinity with modeled spike glycoprotein and the best template protein structure were selected for further investigations. Ensuring the successful passage of a drug through the body is of utmost importance to avoid the failure of those compounds during clinical investigations and to improve the likelihood that they will become drug candidates in the future. These parameters are investigated by the Molsoft L.L.C. (Molecular Properties and Drug-likeness) server (https://molsoft.com/mprop/). The toxicological purpose is the primary consideration in the case of the introduction of new drugs. The toxicity assessment was carried out on the top potential hit compounds using the DataWarrious software [44]. Hydroxychloroquine, lopinavir, ritonavir, and ruxolitnib were used as standards.
Table 1. The docking search space with centres and dimensions 
Results and Discussion
Phylogenetic relationships of the SARS-coronavirus -2 SP
Software called Molecular Evolutionary Genetics Analysis (MEGA) is used to create phylogenetic trees and conduct statistical analyses of molecular evolution. A phylogenetic tree of SARS-coronavirus-2 SP (accession ID: QIG55955.1) and another SARS-coronavirus-2 SP from the NCBI database were constructed using the UPGMA method [30]. The PDB 7CWL and 6XR8 are closely connected to the query protein sequence (accession ID: QIG55955.1) with a bootstrap value of 97%, according to the phylogenetic tree construction (Figure S1). Bootstrap values are considered stable if they are greater than 95%, and unstable or phylogenetic trees will alter when reconstructed if they are less than 70% [45]. In this study, the genetic distance, or scale bar, is 0.0030, indicating a change in the base of 3 nucleotides every 1000 bases. The closer the kinship, the lower the bar scale value [46].
Homology modeling
The phylogenetic analysis for SARS-coronavirus-2 indicated the highest query cover (100%) with spike glycoprotein (SARS-coronavirus-2) (accession number QIG55955.1), suggesting that PDB ID: 7CWL_A was the most suitable template for homology modeling. The dendrogram tree analysis shows that PDB ID 7CWL has the minor resolution of 1.0 Å and is the best template to build the homology modeling (Figure S2). These results supported the phylogenetic tree presented using MEGA X based on the amino acid sequences of the SARS-coronavirus-2 spike glycoprotein (Figure S1). The final alignment was used in the homology modeling of the known sequence with the prototype 3D structure of spike glycoprotein (PDB ID: 7CWL_A), as revealed in Figure 1.


 Figure 1. Sequence alignment results between the query and the template sequence of SARS-Cov-2 SP. The sign (*) denotes identical amino acids
Figure 1. Sequence alignment results between the query and the template sequence of SARS-Cov-2 SP. The sign (*) denotes identical amino acids
Protein structure validation
Figure S3A depicts the 3D structural model of spike glycoprotein. It has an ERRAT overall quality factor of 47.990 (Figure S3B). The 3D structure of the template (PDB: 7CWL) is shown in Figure S4A, and its ERRAT overall quality factor is 63.524 (Figure S4B). The ERRAT overall quality factor suggests that the model is very reliable. The Protein Structure Analysis (ProSA) of modeled protein and the template (PDB: 7CWL) 3D structure give Z-scores of-9.02 and -10.87, respectively, demonstrating that the proteins are in one of the many suitable protein structures discovered experimentally using X-ray crystallography (Figures S5A and S5B).
In Figures S6 and S7, the Ramachandran plot depicts the area of possible angle forms by psi and phi angles. In peptides, the traditional phrase relates to the torsional angles on both sides of the α-carbon. The Ramachandran plot of the modeled SARS-Cov-2 spike glycoprotein structure revealed that 86.5% of the residues were located in the most favorable allowed regions (Figure S6), which was greater than the percentage of residues (85.8%) in the most favorable allowed regions for the initial template (PDB ID: 7CWL spike glycoprotein structure from comparative modeling (Figure S7), clearly showing that the enhanced modeled crystal structure was more stable. The percentage of residues found in further allowed regions, generously allowed regions, and disallowed regions were 11.0%, 1.9%, and 0.7%, respectively. The molecular template structure of Severe acute respiratory spike glycoprotein (PDB ID: 7CWL) only has 12.7%, 1.4%, and 0.1% residues in the further allowed regions, generously allowed regions, and disallowed regions, respectively (Figure S7). This indicates that the modeled SARS-Cov-2 crystal structure ought to be robust and dependable for use in docking simulations to elucidate receptor-ligand binding modes through docking simulations.
Additionally, the structure validation of the modeled protein (Figure S8) and the template protein (Figure S9) was achieved, revealing a pass. The average 3D-1D score for 89% of the residues in the 3D environment profile is 0.2, supporting the veracity of the modeled and template protein crystal structures. Figure S10 depicts the template protein's energy profiles and modeled proteins' energy profiles. The reliability of the spike glycoprotein model forecasted by ProSA-web, SAVES-web, and the DOPE score profile from MODELLER v10.3 (Figure S10C) suggested that this structure used high-quality theoretical protein structure models.
Docking simulation analysis
The docking simulation technique has recently been used in biological studies for pharmaceutical research to determine the precise binding position of proteins and ligands [47]. It estimates the interaction of two compounds with the lowest total energy and the best ligand-receptor orientation [48, 49]. All five compounds underwent a molecular docking investigation against the SARS-coronavirus-2 spike glycoprotein. The binding affinities of all the docked compounds against modeled spike glycoprotein and PDB ID: 7CWl spike glycoprotein are shown in Table 2, which reflects their binding interactions with the receptors. Based on the docking score, it is evident that all the compounds interact better in both protein structures. Due to their improved binding conformations, compounds with low binding affinity are more suited to function as therapeutic molecules. The top two candidates are chosen for additional examination according to their binding affinities. The Ritonavir drug having the least binding affinity, was selected to be taken as a reference. The standard drug (Ritonavir) binding affinity against the modeled and known spike glycoprotein receptors is -6.7 and -7.3 kcal/mol, respectively. We noticed compound 36 has a high affinity against unknown (modeled spike glycoprotein structure) and known (PDB ID: 7CWL) targets.; theoretically, these compound 36 “(S)-N-(4-carbamoylphenyl)-8-cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-2,3-dihydro-5H-thiazolo[3,2-a]pyridine-3-carboxamide” can inhibit those targets and, consequently, their anti-SARS-Cov-2 activity.
Table 2. The affinities of the selected compounds against SARS-Cov-2 spike glycoprotein 
We could see how the two compounds with the highest binding affinities were chosen and interacted with the two probable targets. The type of amino acids present in the active center and their positions and the receptor's two-and three-dimensional structures can affect the binding affinity in receptor-ligand interactions. As a result, various binding affinity profiles were found for each receptor-ligand pair examined in the present study. Figures 2, 3, 4, and 5 display the docked ligand molecules with spike glycoprotein and emphasize each amino acid necessary for ligand-receptor binding. According to these results, compounds 36 and ritonavir showed high binding affinity to target proteins. As opposed to that, hydroxychloroquine was one of the compounds with the worst affinity for all protein targets (Table 2). The predicted binding affinity of compound 36 on the spike glycoprotein was found to be between -8.2 and -8.6 kcal/mol, respectively. The van der Waals interactions show the highest residue interactions in the receptor-ligand interplay. Conventional hydrogen bonds and hydrophobic interactions were also frequently found. In the interaction between ritonavir and the modeled spike glycoprotein, the interaction with the receptor binding site amino acid residues formed a conventional hydrogen bond with THR1117 (4.01 Å), as well as hydrophobic interactions with ILE712 (5.50 Å) and PRO1141 (5.62 Å) (Figure 2). Compound 36 forms two conventional hydrogen bond interactions with ALA706 (3.12 Å and 4.96 Å). Compound 36's rings interact hydrophobically with ALA713 (4.14), LEU1145 (4.43 Å and 4.48 Å), ILE712 (6.68 Å), and PRO1143 (4.69 Å) (Figure 3). The same selected compounds performed very well when docked with the template crystal structure PDB ID: 7CWL, producing the highest binding affinity score. It is quite interesting to note that all the hits produced are on the surface of the crystal structure compared with the modeled receptor. The docking results for the Ritonavir compound show a conventional hydrogen bond with SER373 and ASN343 residues, electrostatic interaction (pi-anion) with ASP364 residue, and one unfavorable donor-donor interaction with ASN439, as shown in Figure 4. While some hydrophobic interactions with the residues VAL367 and LEU368 stabilize the interactions of the “(S)-N-(4-carbamoylphenyl)-8-cyclopropyl-7-(naphthalen-1-ylmethyl)-5-oxo-2,3-dihydro-5H-thiazolo[3,2-a]pyridine-3-carboxamide” (compound 36), compound 36 interactions with amino acids (Figure 5) are very similar to Ritonavir binding interactions. GLY339, PHE338, PHE342, SER371, PHE374, SER374, TRP436, ASN440, SER438, and ASN437 are the common amino residues in the docking sites.
ADMET studies and Lipinski rule of five
To make the medications pertinent in clinical preliminaries, it is essential to concentrate on their retention, dissemination, digestion, discharge, and poisonousness in the human body before beginning the examination conventions [50], regarding a few important principles, for example, those of Lipinski's five [51, 52] and Veber's standard [53]. This method also excludes molecules with potentially unfavorable physiological features, such as toxicity and pharmacokinetic properties [54]. We used the online MOLSOFT (https://molsoft.com/mprop/) and DataWarrior [44] software to assess the drug similarity and in silico pharmacokinetic features of the two selected drugs as SARS-Cov-2 spike glycoprotein inhibiting agents. The results in Table 3 demonstrate that compound 36 has adequate pharmacological properties for ritonavir drug. According to the results, compound 36 meets the requirements. However, the molecular weight of a drug plays a significant role in its oral bioavailability.
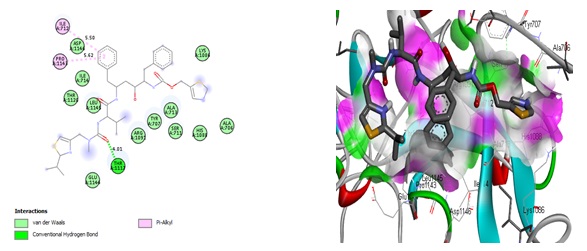
Figure 2. Interactions of Ritonavir and the modeled SARS-Cov-2 spike glycoprotein residues
 Figure 3. 2D and 3D interactions of Ritonavir and the modeled SARS-Cov-2 spike glycoprotein residues
Figure 3. 2D and 3D interactions of Ritonavir and the modeled SARS-Cov-2 spike glycoprotein residues
 Figure 4. 2D and 3D interactions of Ritonavir drug at the binding site of the 7CWL protein
Figure 4. 2D and 3D interactions of Ritonavir drug at the binding site of the 7CWL protein
 Figure 5. 2D and 3D interactions of compound 36 at the binding site of the 7CWL protein
Figure 5. 2D and 3D interactions of compound 36 at the binding site of the 7CWL protein
The 500 Da cut-off does not differentiate between poor or high oral bioavailability drugs. The table's permeability data reveal that compound 36 does not cross the BBB. Furthermore, compounds that do not meet three requirements of Lipinski’s rule of five are qualified and are not orally bioavailable. Ritonavir violated three of Lipinski’s rules, e.g., the molecular weight is higher than 500, the number of hydrogen bond donors is higher than 5, and molLogP is higher than 5. Drugs with high intestinal absorption, distribution (action), and oral bioavailability have high MolLogS, Number of Rotatable Bonds (NRB), and MolVol values. Both compounds having low aqueous solubility level are shown in Table 3, and compound 36 have shown moderate solubility. The properties of a compound's absorption and distribution are greatly influenced by its aqueous solubility (MolLogS). Slow absorption and action result from limited solubility. Veber's rule states that MolPSA must be less than or equal to 140 Å2, and the number of rotatable bonds must be less than or equal to 10. It was observed that the Ritonavir drug violated Veber’s rule, as NRB is equal to 18. DataWarrior v5.5.0 determined the mutagenic, tumorigenic, reproductive effects, and irritant levels of compounds. It stipulated that certain functional groups, mutagenic, tumorigenic, reproductive effects, and irritant properties, could influence toxicity. Table 3 reveals that compound 36 tends to be tumorigenic, while the standard drug (Ritonavir) has none of the toxicity risk factors. However, when the ADMET results were compared with a drug-likeness model score in Figure 6, compound 36 could be a drug (Figure 6A). In contrast, Ritonavir has less potential according to the drug-likeness model score (Figure 6B).
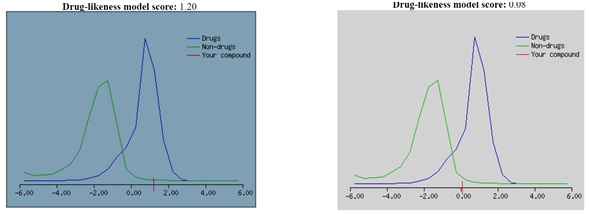
Figure 6. Drug-likeness model score for (A) compound 36 and (B) Ritonavir drug
Table 3. Pharmacokinetic properties of the selected compounds obtained from the docking results

Conclusions
In the present work, we studied five compounds using phylogenetic analysis, homology modeling, docking simulations, and ADMET studies. In the phylogenetic analysis, SARS-Cov-2 spike glycoproteins were divided into 3 clades based on the analysis of the spike glycoprotein sequence. The SARS-coronavirus-2 spike glycoprotein sequences 7cwl, 6xr8, 7krq, and 6zwv` were grouped with the query sequence accession number QIG55955.1. The sub-group is 7cwl, 6xr8, and the query sequence is closely related. The dendrogram tree analysis from the MODELLER revealed the importance of 7cwl.pdb for homology modeling, which guided us to model a new spike glycoprotein crystal structure. The docking studies executed with all the compounds and compound 36 and the reference drug Ritonavir proved to be the most active compounds. The docking simulation reveals that the most active compound 36, forms significant connections with the active sites as it was discovered with known inhibitors of the modeled and PDB 7cwl forms of SARS-Cov-2 spike glycoprotein. Lipinski's rules and drug-likeness properties were predicted for compound 36 and Ritonavir. The predicted values of compound 36 were in good agreement with drug-likeness characteristics. As a result, the pharmacokinetic properties and docked poses reveal details of the expected binding modes and essential chemical interactions, which may allow pharmaceutical companies and biomedical chemists to build new Covid-19 medicines.
Acknowledgment
The authors gratefully acknowledged the technical effort of Adawara Samuel Ndaghiya, Department of Pure and Applied Chemistry, University of Maiduguri, Borno State, Nigeria.
Disclosure statement
The authors reported no potential conflict of interest.
ORCID
E.I. Edache : 0000-0002-2976-9636
A. Uzairu : 0000-0002-6973-6361
)