Document Type : Original Research Article
Authors
Department of Chemistry, Faculty of Physical Sciences, Ahmadu Bello University, P.M.B.1045, Zaria, Kaduna State, Nigeria
Abstract
Leishmaniasis, a neglected tropical disease, affects a human population of over 12 million people, mainly in the tropics, and is caused by the leishmanial parasites transmitted by the female sand fly. The lack of vaccines to prevent leishmaniasis, and the limitations of existing therapies necessitated this study, which was focused on the computational design of some novel 2-arylbenzimidazole analogs while subjecting the same to molecular docking, pharmacokinetics, and Molecular Dynamic (MD) simulation to ascertain their drug-ability. The obtained QSAR model was validated as follows: R2 = 0.8447, R2adj = 0.8095, Q2cv = 0.6799, and R2test = 0.7383. The predicted pIC50 values of all newly designed compounds were higher than that of the lead compound (13). The newly designed analogs conformed to Lipinski's rule for oral bio-availability less 13m, and showed good ADMET properties, with 13j showing the highest Human Intestinal Absorption (HIA) of 93.64%. The MD simulation result demonstrated excellent stability associated with the binding of 13j onto 6K91's binding pocket with an estimated ∆G binding (MM/GBSA) of -50.4031 kcal/mol than that of 13_6K91 (∆G binding = -25.9665 kcal/mol). Therefore, 13j could be recommended as a potential drug molecule for treating leishmanial infections.
Graphical Abstract
)
Keywords
Introduction
Leishmaniasis is a tropical disease that infects approximately 12 million people living majorly in tropical Africa, Southeast Asia, and Latin America [1]. Visceral leishmaniasis (VL) is the deadliest of all leishmanial infections and always proves fatal if left untreated. The leishmanial (L) parasites cause VL, majorly Leishmania donovani and Leishmania infantum, which are spread to humans by sand flies [2]. Significant health challenges associated with leishmaniasis include weakness, weight loss, fever, and hepatosplenomegaly [3]. Because of a lack of a vaccine to prevent this infection, a treatment approach has been chemotherapy with drugs such as amphotericin B, pentamidine, paromomycin, and miltefosine, among others [4]. However, these therapies are either ineffective or are associated with adverse effects such as nausea, hepatotoxicity, nephrotoxicity, eye irritation, lethargy, and cardiotoxicity [5,6]. More so, resistance posed by target organisms to existing therapies is on the rise [7]. Additionally, leishmaniasis is taken less seriously than other diseases like cancer, diabetes, malaria, stroke, hepatitis, and AIDS [8]. Hence, efforts geared toward discovering and developing new medicines that overcome the limitations of existing therapies are necessary.
Computer-aided modeling approaches such as DFT calculations, QSAR modeling, molecular docking, drug-likeness properties prediction, molecular dynamics (MD) simulation, and homology modeling play a critical role in drug discovery and development owing to their advantages over traditional methods such as time-saving, cost-effective, and reliability [9-12]. The pyridoxal kinase (PdxK) protein (PDB: 6K91) was reported previously to play a vital role in the formation of pyridoxal -5’-phosphate, an active form of vitamin B6 by catalyzing the phosphorylation of the 5’-hydroxyl group of the pyridoxal [13]. PdxK is a crucial enzyme for the growth of parasites and is also valuable for host infection [14]. PdxK has previously been inhibited by well-known anti-malarial medicines such as primaquine and chloroquine [14]. Therefore, PdxK could serve as an exciting enzyme target for new leishmanial inhibitors.
The benzimidazole structure is found in several drugs used to treat various medical conditions [15]. Additionally, benzimidazole-derived compounds have been previously studied as leishmanial inhibitors [16-18]. In the continuous search for an ideal anti-leishmanial drug compound, Keurulainen et al. [15] synthesized a series of 2-arylbenzimidazole derivatives and evaluated their activities against L. donovani. Therefore, the present study focuses on 2-D QSAR modeling, in silico design, molecular docking investigation, pharmacokinetics profiling, and molecular dynamics studies to identify some 2-arylbenzimidazole derivatives as superior leishmanial inhibitors.
Materials and Methods
Data acquisition
Keurulainen et al. [15] synthesized a series of 2-arylbenzimidazole derivatives, contributing to the drug discovery stride to tackle leishmaniasis. Their bioactivities were evaluated against axenic amastigotes of L. donovani at 50 µM concentration and reported as percent (%) inhibition. Consequently, a dataset of Forty (40) compounds with relatively better inhibition values was obtained from their report and used for this theoretical study. The various % inhibition values were converted to half-maximal inhibitory concentration on a logarithmic scale (pIC50) using Equation 1. The molecular structures, % inhibition, and pIC50 obtained for these compounds were reported in the supplementary file Table S1.
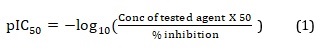
Structural optimization
The ChemDraw Ultra software was used to draw the molecular structures of the various analogs and after saved in MDL molfile. The resulting files were opened separately on the Spartan ’14 user interface and converted from 2-D to 3-D models. The resulting structures were subjected to a two-step optimization process; first with Molecular Mechanics Force Field (MMFF) and after that with Density Functional Theory (DFT) with B3LYP/6-31G** basis set, all on Spartan 14 software version 1.1.4 [4,6,8]. The resulting structures were then preserved in an SD file for use in the generation of molecular descriptors [10,19].
2-D QSAR model building
The PaDEL-Descriptor tool generated a pool of molecular descriptors for all Forty (40) compounds [10]. DTC-Lab-based pretreatment software, GUI 1.2 was used to treat and screen the data of uninformative descriptors [9]. The pretreated data were divided into a training set and a test set in a 70:30 proportion [20]. The actual model building was carried out on the Material studio software, which employs the Genetic Function Approximation (GFA) and Multi-Linear Regression (MLR) approach to build the QSAR model. MLR provides the relationship between the inhibitory activity (pIC50) and the molecular descriptors [21].
The next important step after the model building is internal and external validation assessment. Some internal validation parameters utilized in this study include correlation coefficient (R2), cross-validation coefficient (Q2cv), and adjusted correlation coefficient (R2adj). An external validation assessment was carried out to ascertain the built model's ability to predict the activities of the external test set compounds. The predictive strength of the QSAR model is dependent on the external test set correlation coefficient (R2 test) [22-25]. In addition, statistical parameters such as the Mean Effect (ME) and Variance Inflation Factor (VIF) were computed to further describe the built QSAR model. The ME value shows the contribution of each descriptor in the model in terms of anti-proliferative activity [26].
In contrast, the level of inter-correlation between the descriptors is defined by the VIF [27]. Furthermore, the model’s Applicability Domain (AD) space was described using the leverage approach [28,29]. Table 1 reveals the various parameters and equations utilized in the model's validation.
Ligand-based drug design
Thirteen (13) new analogs of 2-arylbenzimidazole were designed by the method of ligand-based drug design basically by the inclusion of relevant substituent group(s) into the structural template (13) using the chemical information encoded in the molecular descriptors (Majorly SpMin1_Bhv and SpMin2_Bhe) [9]. The newly designed analogs and the reference drug (pentamidine) were prepared according to the procedures earlier described herein.
Molecular docking study
Pyridoxal kinase (PdxK), the target receptor (PDB ID: 6K91) was obtained from the protein data bank and prepared using the Biovia Discovery Studio by removing water molecules and co-crystallized ligands associated with the protein structure. A molecular docking investigation was performed between PdxK and the 13 newly designed analogs using the PyRx software (Auto Dock Vina tool). This was to ascertain how well the new compounds interact with the receptor of interest. Pentamidine was also docked onto the same binding pockets of PdxK to revalidate the docking results. The vina search space was centered at X: 29.3648Å, Y: 1.3484Å, and Z: 27.9440Å with dimensions X: 50.6424Å, Y: 46.7480Å, and Z: 54.4647Å [22,26].
Table 1. Selected parameters and equations used for the QSAR model's validation

Pharmacokinetics study
Drug-likeness and ADMET properties prediction are necessary for the initial stage of drug discovery because only molecules with good pharmacokinetic profiles scale into the pre-clinical phase of drug research [10]. In the present study, the pharmacokinetic properties of the template molecule (13) and the newly designed compounds (13a–13m) were predicted by using http://www.swissadme.ch/index.php and http://biosig.unimelb.edu.au/pkcsm for drug-likeness and ADMET profiling respectively. Lipinski's ‘rule of five’ (ROF), a widely used criterion for oral bioavailability, was used to assess the newly designed compounds for oral bioavailability [30, 31].
Molecular dynamics study and calculation of MM/GBSA
MD simulations were performed on the complexes of compound 13 (template) and compound 13j with PdxK using the combined approach of Chemistry at Harvard Macromolecular Mechanics (CHARMM) force field, Nano-scale Molecular Dynamics (NAMD), and Visual Molecular Dynamics (VMD). The CHARMM-GUI, an established web-based platform that utilizes the CHARMM force field, was used to generate the input files for the simulation by NAMD [32]. The periodic boundary condition was utilized while fitting the system into a cubic water box for solvation. The protein was solvated and neutralized explicitly in an aqueous solution of 0.15 M concentration of potassium chloride salt [33]. The simulation process involving energy minimization, equilibration (100 ps time frame), and production (500,000 steps or 1 ns time frame) was performed on the resulting system. At the same time, the results were visualized using VMD and the Biovia discovery studio [34]. A similar procedure was described elsewhere [33]. Additionally, MolAICal software was used to compute the ligand-binding affinity by Molecular Mechanics Generalized Born Surface Area (MM/GBSA) method based on the resulting MD log files obtained with NAMD [35]. MM/GBSA is estimated using Equation (11)–(13) [35].
![]()
![]()
![]()
Where, ∆EMM is the gas phase MM energy and -T∆S represents conformational entropy. ∆EMM contains electrostatic ∆Eele, ∆Evdw, and ∆Einternal of bond, angle, and dihedral energies. At the same time, ∆Gsol is the solvation-free energy equal to the sum of the nonelectrostatic solvation component ∆GSA and electrostatic solvation energy ∆GGB.
Results and discussion
QSAR modeling
A 2-D QSAR study was carried out on Forty (40) 2-arylbenzimidazole derivatives, to establish a quantitative relationship between their structures and anti-leishmanial activities. The built model (Equation 14) was assessed for internal and external validation tests and was found to satisfy the requirement for a good QSAR model. The various descriptors used in the QSAR model were described in Table 2, while the observed and predicted activity values and their residual values, were presented in Table 3. Internal and external validation tests were carried out to ascertain the stability, robustness, reliability, and predictive strength of the built QSAR model, and the results are presented in Table 4.
Table 2. Selected molecular descriptors used in the QSAR model building

Table 3. Observed, predicted and residual values of 2-arylbenzimidazole derivatives


A combined GFA and MLR approach led to the selection of (5) descriptors, and generation of Four (4) QSAR models respectively. Model 1 (Equation 14) was found to satisfy the requirement for a reliable QSAR model. The low residuals between the observed and predicted activities as shown in Table 3 connote a high predictive strength of the model.

Table 4. Validated parameters of the QSAR model

Another critical analysis performed on the five descriptors was Pearson’s correlation statistical analyses, with the results shown in Table 5. The low values of coefficients of correlation (< 0.50) that exist between any pair of descriptors in the model (Table 5) connote no relationship between each descriptor. The VIF values for the various descriptors range between 1 and 5, showing the built model's stability and acceptability (Table 5). The selected descriptors were good, as shown by the values of absolute t-statistics greater than 2 [36]. Also available in Table 5 were the evaluated p-values at a 95% confidence level for all descriptors, which were less than 0.05. A similar trend in statistical parameters was obtained elsewhere for QSAR modeling of maleimides [8] and arylimidamide-azole hybrids [4] as anti-leishmanial agents. Therefore, the alternative hypothesis states that a relationship exists between inhibitory activities and the descriptors holds.
Furthermore, the Mean Effect (ME) values reported in Table 2 revealed crucial information on the contributions of each descriptor in the built model. The magnitudes and signs of ME values signify their respective strength and direction on the molecules’ inhibitory activities. The various descriptors except SpMin2_Bhe have negative ME, meaning that an increase or decrease in their values will result in a corresponding decrease or increase in the inhibitory activities respectively. On the other hand, an increasing value of SpMin2_Bhe will lead to an increase in anti-leishmanial activities. SpMin2_Bhe has the most significant influence on the molecules’ inhibitory activities because it has the highest ME value of 3.88. SpMin2_Bhe, the smallest eigenvalue n. 2 of the Burden matrix weighted by Sanderson electronegativity, was suggested to contribute positively to anti-leishmanial activity. It means that introducing an electronegative substituent or group will lead to an increase in the anti-leishmanial activity [4].
Table 5. Descriptors’ inter-correlation and statistical analyses of the built QSAR model

The plot of the standardized residuals against the leverages (William’s plot) was obtained to determine the model’s applicability domain (Figure 1). It was observed from the plot that all the molecules fall within the square area of ±2.5. Hence, no outlier was present in the data set. However, five compounds (1, 6, 16, 36, and 37) were found beyond the calculated cut-off leverage (h* = 0.64) and were said to be influential molecules.
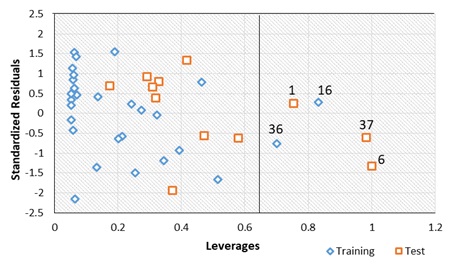
Figure 1. The William’s plot
Consequently, compound 13, with a relatively higher predicted activity of 4.6235 (Table 3), has also been found within the model's applicability domain space (Figure 1) and, with low residual value, was chosen as a template molecule for designing new prominent inhibitors.
Ligand-based drug design
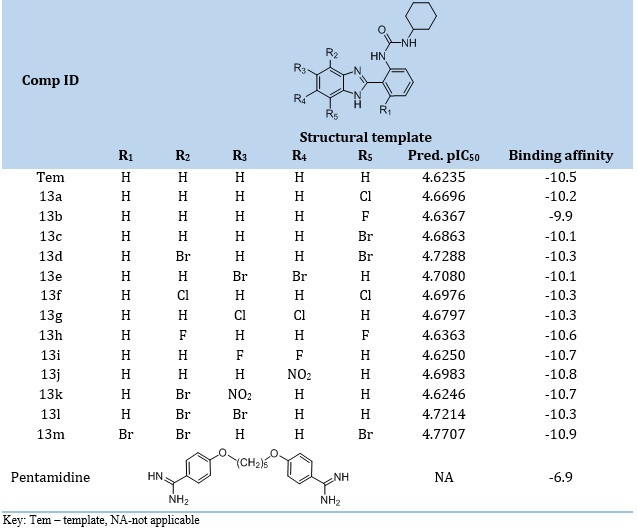
One of the main objectives of the ligand-based design is designing new inhibitors with better inhibitory activities compared to that of the template compound used for the design. The structural template, molecular structures, predicted activities, and binding affinities (kcal/mol) of the new analogs and pentamidine are shown in Table 6. In the present study, the predicted pIC50 values of the 13 newly designed analogs (13a – 13m) were higher than that of the template molecule (13), with 13m and 13k having the highest and lowest predicted pIC50, respectively (Table 6). It, therefore, affirmed that the various structural modifications of the template structure yielded the desired result as it was based on the information encoded by the molecular descriptors in the built model.
Binding affinities of protein-ligand interactions are necessary to describe how strong the ligand-receptor binding is. The spontaneity of the binding process is shown by the negative value of the binding affinity [10]. Here, the binding affinities of the newly designed compound range from -9.90 kcal/mol to -10.90 kcal/mol and are higher than that of the reference compound, pentamidine (-6.90 kcal/mol). However, only 13h, 13i, 13j, 13k, and 13m possessed higher binding affinities than the template molecule (-10.50 kcal/mol).
Table 6. Molecular structures predicted activities and binding affinities of the new analogs
Pharmacokinetics properties prediction
The results of the pharmacokinetics investigation conducted on the thirteen (13) newly designed compounds were presented in the supplementary file Table S2 – S3. At the same time, Figure S1 (a-b) shows the oral bioavailability radar of 13 and 13j.
Lipinski’s approach to ascertaining the oral bioavailability of compounds has been widely applied in discovering new drug molecules [10]. It states that an orally bioavailable drug molecule is likely to have an HBD of less than 5, HBA < 10, MW < 500, and MLOGP < 4.15 or WLOGP < 5 [31]. A molecule is said to obey the ROF for oral bioavailability when it passes at least three of the four provisions of the rule [37]. As observed in Table S2, the template and all the newly designed analogs passed the drug-likeness test (Lipinski RO5) except 13m which failed two of the rules (MW and MLOGP of greater than 500 and 4.15, respectively). The reported values of Topological Polar Surface Area (TPSA) for the molecules were below the threshold value of 140 Å2, above which a molecule tends to show poor Human Intestinal Absorption (HIA). Also, the synthetic accessibility (SA) values of these compounds were less than 5.00 (easy portion on a scale of 1 to 10), suggesting easy laboratory synthesis of these molecules. A similar observation was reported elsewhere for drug-likeness analysis of designed pyrazolopyrimidine analogs [6]. The estimated water solubility (Log S) of the various molecules suggests moderate solubility for the new analogs except 13d, 13e, 13l, and 13m which were predicted to be poorly soluble in aqueous media.
The estimated ADMET properties reported in Table S3 showed an excellent Human Intestinal Absorption (HIA) (greater than 75 %) for all tested compounds, with 13j showing 93.64% HIA. Skin permeation is crucial in transdermal drug delivery development, where the skin permeation constant (LogKp) of greater than -2.50 connotes poor skin penetration. Consequently, the various tested compounds showed LogKp values of less than -2.50, indicating good skin permeability. For a drug molecule to penetrate the Blood-Brain Barrier (BBB) and Central Nervous System (CNS) readily, the logarithmic ratio of brain to plasma drug concentration (logBB) must be greater than 0.3, and the blood-brain permeability-surface area product (logPS) be less than -2 respectively. Consequently, none of these molecules were predicted to readily penetrate the BBB. However, all the tested molecules were predicted to permeate the CNS except 13h and 13i.
Furthermore, the cytochrome P450 enzymes are necessary for the body to aid drug metabolism and facilitate excretions. The two essential isoforms enhancing drug metabolism, CYP-34A, and CYP-2D6, were tested. The tested molecules were not substrates and inhibitors of the CYP-34A enzyme. However, all analogs were substrates of CYP-2D6 except the template compound (13), an indication that the metabolic process of the template in the system may be slow. Also, all tested ligands were substrates of P-glycoprotein, an enzyme that acts as a biological barrier by extruding toxins and xenobiotics, including drugs, out of cells.
Interestingly, all the analogs showed inhibition to this enzyme except 13i. This means that these analogs, when taken into the human system, may likely not be effluated out of the target cells by this enzyme. The drug's total clearance ascertains the extent of drug elimination from the body. The range of total clearance values for all the tested molecules is good.
Additionally, the predicted values of Maximum Recommended Tolerated Dose (MRTD) for all tested molecules were reported in Table S3. MRTD ≤ 0.477 log (mg/kg/day) is considered low and high if > 0.477 log (mg/kg/day). The overall drug-likeness and ADMET properties revealed excellent pharmacokinetic profiles for these molecules except for the analogs predicted to be poorly soluble (13d, 13e, 13l, and 13m).
Pharmacological interaction study
The pharmacological interactions between the receptors’ amino acid residues and the Three (3) compounds of 13 (Template), 13j (designed analog), and reference compound (pentamidine) before and after MD simulations were summarized in the supplementary file Table S4, while the binding interactions of 13 and 13j with PdxK after MD simulations were shown in Figures 2 and 3 respectively. This provided insight into the mode of binding of these ligands with the active sites of the target protein (PdxK). The choice of 13j (pIC50 = 4.6983) over 13d (pIC50 = 4.7288), 13e (pIC50 = 4.7080), 13l (pIC50 = 4.7214), and 13m (pIC50 = 4.7707) was based on its excellent pharmacokinetic properties.
The pharmacological interactions between the receptors’ amino acid residues and the Three (3) compounds of 13 (Template), 13j (designed analog), and reference compound (pentamidine) before and after MD simulations were summarized in the supplementary file Table S4, while the binding interactions of 13 and 13j with PdxK after MD simulations were shown in Figures 2 and 3 respectively. This provided insight into the mode of binding of these ligands with the active sites of the target protein (PdxK). The choice of 13j (pIC50 = 4.6983) over 13d (pIC50 = 4.7288), 13e (pIC50 = 4.7080), 13l (pIC50 = 4.7214), and 13m (pIC50 = 4.7707) was based on its excellent pharmacokinetic properties.
The various compounds were said to interact very adequately with the target receptor as shown by the presence of hydrogen bonding (H-bond), hydrophobic interactions, and in some cases electrostatic interactions (Table S4). The binding profile of compound 13j with PdxK involved a total of Three (3) conventional H-bonds, One (1) C-H bond, One (1) π-anion electrostatic interaction, and up to Four (4) hydrophobic interactions. The presence of the nitro group in 13j played a significant role in forming 3 conventional H-bonds with ASN-151, THR-229, and GLY-230 at interaction distances of 2.17Å, 1.85Å, and 2.43Å respectively. The carbonyl group (C=O) oxygen in 13j formed a C-H bond with THR-227 at 3.78Å. Also visible was the π-anion interaction between the π-electrons systems of ASP-231 and the five-membered ring system of the benzimidazole group at 3.95Å. Others include hydrophobic interactions such as π-π stacked with TYR-85 (4.84Å), alkyl with ARG-225 (4.99Å), and π-alkyl with VAL-19 (4.04Å) and VAL-121 (4.89Å).
Regarding the number of interactions formed, the binding interaction pattern of the simulated complex 13_6K91 (Figure 2) deviated only slightly from that of the non-simulated complex (Table S4). However, a significant improvement in H-bonding interactions was observed in the case of simulated complex 13j_6K91 (Figure 3) over the non-simulated complex (Table S4) as Seven (7) conventional H-bonds, Two (2) C-H bonds, and One (1) π-donor H-bond were formed. This indicates more stable interactions as the compound interacts with the target in a dynamic system. It is important to note that the real system in which drugs interact with receptors is dynamic. Hence, it became imperative to examine how the selected ligand-protein complex behaves in a dynamically simulated system, which can be likened to the real drug-receptor system.

Figure 2. Binding interaction between 13 and Pyridoxal kinase (PDB: 6K91) after MD simulation
 Figure 3. Binding interaction between 13j and Pyridoxal kinase (PDB: 6K91) after MD simulation
Figure 3. Binding interaction between 13j and Pyridoxal kinase (PDB: 6K91) after MD simulation
Molecular dynamics study and calculation of MM/GBSA
To ascertain the stability and rigidity of the protein-ligand interactions, the complexes of compound 13 (template) and compound 13j were subjected to MD simulation. The results of the simulation are summarized as plots of Root-Mean-Square Deviation (RMSD), Root-Mean-Square Fluctuation (RMSF), Solvent Accessible Surface Area (SASA), and Radius of gyration (Rg) versus the time in picoseconds (ps), and presented in supplementary file Figures S2-S5. The average RMSD values were estimated as 2.74078Å and 2.53116Å for 13_6K91 and 13j_6K91, respectively, which showed that 13j_6K91 deviated slightly less than 13_6K91 (Figure S2). An indication that 13j_6K91 is more stable [25]. RMSF is more like a calculation of the flexibility or the extent of movement of individual residue during a simulation. The RMSF plot in Figure S3 showed that the flexibilities of the protein residues were almost constant during the trajectory, an indication of the stability of the interactions during the simulation. The SASA is simply the surface area in contact with the complex's solvent. From Figure S4, it can be observed that the SASA lay between 13500 Å2 and 15500 Å2 for 13_6K91 and between 13500 Å2 and 14500 Å2 for 13j_6K91 during the trajectory. The Rg measures the degree of protein’s compactness during the trajectory. Decreasing Rg indicates reducing residues’ flexibilities and more stability for the protein. Throughout the trajectory, the Rg varies between 18.05 Å and 18.50 Å, equivalent to a difference of less than 0.5 Å for the complexes studied, connoting slight changes in the protein compactness as the simulation progresses, means the stability of the complexes (Figure S5). Furthermore, the result of binding free energy (MM/GBSA) computed for 13_6K91 and 13j_6K91 by MolAICal is presented in Table 7.
Table 7. Binding free energy parameters of 13_6K91 and 13j_6K91 complexes

The negative value of the estimated binding free energy (MM/GBSA) for both complexes (-25.9665 kcal/mol and -50.4031 kcal/mol for 13_6K91 and 13j_6K91 respectively) indicates the favorability of the ligand-protein binding. A similar result was obtained elsewhere for a newly designed pyrazolopyrimidine analog [38]. The binding free energy of 13j_6K91 is approximately twice that of 13_6K91, indicating that 13j_6K91 could be more stable than 13_6K91. It can therefore be inferred that compound 13j binds more readily with the pyridoxal kinase even within a dynamically perturbed system. It hence could be considered a potential drug candidate for treating leishmanial infections.
Conclusions
The present study developed a QSAR model with a series of Forty (40) arylbenzimidazole derivatives as anti-leishmanial agents, which satisfied the criteria for internal and external validation assessments. The built model was used to predict the anti-leishmanial activities of the various analogs, including the newly designed compounds. A combined molecular docking and pharmacokinetic screening identified one of the newly designed analogs (13j) as a promising pyridoxal kinase inhibitor. Results of the MD simulation showed the favorability of the binding interactions between the template (13) and the receptor (PdxK), as well as between 13j and PdxK, with 13j_6K91 being more energetically stable (∆G binding = -50.4031 kcal/mol) than 13_6K91 (∆G binding = -25.9665 kcal/mol). The newly designed analog (13j) appeared to be more consistent with the various validation protocols employed in this study and, therefore, could be recommended as a potential drug molecule for treating leishmanial infections.
Acknowledgment
The authors sincerely acknowledge G.F.S Harrison Quantum Chemistry Research Group, Ahmadu Bello University Zaria, for their guidance and support during this work.
Disclosure statement
The authors reported no potential conflict of interest.
ORCID
Fabian A. Ugbe : 0000-0001-7052-4959
Gideon A. Shallangwa : 0000-0002-0700-9898
Adamu Uzairu : 0000-0002-6973-6361
Ibrahim Abdulkadir : 0000-0002-4025-1889
)