Document Type : Original Research Article
Authors
Department of Chemistry, Government Arts College (Autonomous), Affiliated to Periyar University, Salem, Tamil Nadu, India, 636007
Abstract
A succinct total synthesis of an indoloquinoline alkaloid was achieved through a one-pot, two-step process involving a cascade of sodium hydride and aniline, followed by a base-mediated Claisen condensation reaction. Subsequently, the reaction was conducted via microwave-assisted synthesis (MAS), resulting in a good yield of the title compound with eco-friendly methodologies. The structural properties of the synthesized compound were validated using FT-IR, 1H-NMR, 13C-NMR, and GC-Mass spectral analyses. In addition, FT-IR approaches were employed to compare the vibrational wavenumbers obtained through simulation with experimental wave numbers. Furthermore, DFT calculations were employed to fine-tune the structural parameters and investigate FMOs, Mulliken atomic charges, MEP, and NLO properties, and molecular docking studies evaluated its N3 binding site of the primary coronavirus protein 6LU7. Likewise, the title compound is assessed for its ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties, revealing promising pharmacokinetic profiles devoid of any detected indications of non-toxicity.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
The indoloquinoline alkaloids, which have a quinoline and indole fused system, are important [1-3]. Notable members of this class include the linear indoloquinoline alkaloid cryptolepine (1) [4,5], cryptotackieine (2), nor-crytotackieine (3), and the angularly fused alkaloid cryptosanguinolentine (4). In addition, isoneocryptolepine (5) was also classified within this group (Figure 1) [6-8]. Isoneocryptolepine alkaloid is derived from a plant of Cryptolepis sanguinolenta. Given its designation, isoneocryptolepine also has antiplasmodial properties and shares several biological characteristics [9]. These include antimicrobial effects, cytotoxicity and DNA intercalation [9]. Cryptolepine and iso-cryptolepine have a traditional application in addressing a wide range of health issues, being employed in clinical therapy for conditions including rheumatism, urinary tract infections of anti-bacterial [11], and malaria diseases [12-14]. The characterization of the designated compound involved the optimization of its structure using DFT at the B3LYP/6.311G* method, focusing on 7H-indolo[2,3-c]quinolinol (IQOL). Subsequently, theoretical investigations into the compound's reactivity were carried out. The investigation integrated both experimental and DFT approaches for FT-IR [15–20].
The synthesis and analysis of indolequinone moieties have been undertaken due to their anti-coronavirus-19 (COVID-19) properties [19], employing computational methodologies to expedite.
The identification and development of innovative therapeutic drug molecules. This method utilizes computational approaches and initial insights from SAR investigations. The results of these studies indicate that optimization may be achieved through precise adjustments to the nature of the substituents on indoloquinoline derivatives [22–24]. Assessing the ADME-Tox analysis of a drug molecule is crucial for researching new drug formulations [25–27].
Recently, chloroquine and hydroxychloroquine have gained prominence as significant medical treatments for COVID-19, ranking among the most prescribed medications in the USA, with more than ten million recommendations. Quinoline alkaloids, a category of compounds used in treating malaria fever infection, have shown efficacy in inhibiting viral replication in diverse infections, including those induced by the SARS-associated coronavirus (CoV) and MERS-CoV. The quinoline heterocyclic structure has proven to be a robust approach in medicinal chemistry research. The exploration of organic synthesis through one-pot, tandem Claisen research in organic chemistry, notably propelling
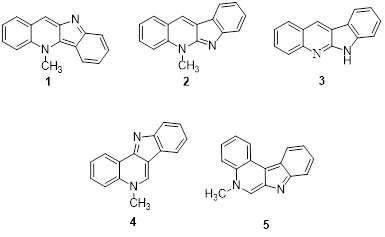
Figure 1. Some selective family of indoloquinoline alkaloids.
drug discovery initiatives. The Claisen condensation reaction is a classical and valuable multicomponent reaction that facilitates the assembly of pharmacologically active quinoline and indole derivatives from commercially available precursors. This advancement holds promise for accelerating drug discovery programs in the field of organic chemistry [28,29].
We devised and synthesized the drug discovery (IQOL)), capable of systematically studying and completing the synthesis of a distinctive compound using computer-generated tools. IQOL has effective inhibitory effects on COVID-19. Furthermore, in silico molecular docking analysis were conducted with the main protein structures of covid-19 (PDB: 6LU7) [29,30]. The approach employed a structure-assisted drug design [32,33]. The primary focus of this program was to pinpoint drug leads targeting the SARS-CoV-2 protein. Finally, ADME/Tox studies [34–37] and [38].
Experimental
Synthesis of 3-hydroxyindole-2-carboxylic acid
Scheme 1 by employing two different routes. In Route A, an equimolar mixture of 1-chlorobenzoic acid 1 (1.57 g, 0.01 mol) and glycine 2 (0.76 g, 0.1 mol), along with (0.50 g) of sodium hydride was added. The resultant mixture underwent microwave radiation at 240 W for 10 minutes.
For Route B, an 0.01 equimolar mixture of ethyl bromoacetate 3 (1.65 g) and methylantharanate (1.6 mL), along with (0.50 g) of sodium hydride, was employed, and then the reaction mixture at 240 W in a microwave oven, 5-10 minutes, The completion of the reaction was confirmed through TLC. It was subsequently purified through column chromatography. The final step included recrystallization from ethanol, leading to a satisfactory 71% yield of the desired product.
FT-IR (KBr, νmax, cm-1): (Figure S1) 3444, 3389, 3048, 1702, 1603, 1445, 1356; 1299, 1188, 881, and 740. 1H-NMR (400 MHz, DMSO-d6): (Figure S2) δ 12.94 (bs, 1OH, C2-COOH), 12.78 (s, H, N-H), 7.96 (dd, J = 8.0, indole – C4H and HC7), 7.62 (t, indole HC6), and 7.50 (t, HC5), 13C-NMR (100 MHz, DMSO-d6) (Figure S3) δ = 167.79, 133.30, 133.03, 131.24, 129.94, 129.72, 129.52, 129.18, 129.01. HRMS (ESI, m/z), (Figure S4) C9H7NO3 [M+H]. = 177.68.
Preparation of IQOL
The mixture comprising equimolar quantities of 3-hydroxyindole-2-carboxylic acid 5 (17.7 g, 0.1 mol) and aniline (9.0 mL, 0.1 mol), along with (0.50 g) of ZnCl2, was introduced [39]. The reaction proceeded under microwave oven conditions at 250 W (180 °C) for 10-15 minutes. A yellow solid was produced, the validity of the reaction procedure was verified through TLC. When the reaction mixture attained room temperature, it was transferred to 500 mL of ice water and neutralized. The formed solid precipitate was filtered, subsequently purified through column chromatography, and subjected to recrystallization from ethanol. resulting in the formation of a yellow solid 63% yield.
FT-IR (KBr, νmax, cm-1): (Figure S5) 3487, 3319, 3129, 3073, 1675, 1594, 1496, 1291, 1182, 816, and 754. 1H NMR (400 MHz, DMSO-d6): (Figure S6) δ 16.22 (bs, OH), 11.52 (s, 1H-N), 7.87 (d, J = 8.4, indole HC11), 7.54 (dd, J = 8.0, quinoline C5-H), 7.50-7.41 (q, C2-H and C8-H), 7.30 (t, H C3), 7.10 (t, HC4), 7.05 (t, 1H, HC9), and 6.89 (t, 1H, HC10). 13C-NMR (100 MHz, DMSO-d6) (Figure S7) δ = 162.51, 157.51, 139.05, 138.09, 137.61, 133.60, 132.69, 129.53, 129.33, 128.58, 127.82, 125.99, 125.76, 125.43, 122.01, 121.43, 120.49, 119.82, 111.75, 109.98, and 106.74; HRMS (ESI, m/z) (Figure S8); C15H10N2O [M-H]. = 233.33.
DFT controls
Quantum mechanism simulations were carried out with the Gauss view and Gaussian 09 W programme. Becke's three-parameter hybrid exchange-correlation functional (B3LYP) with a 6-311G (d,p) basis set was used for these computations. Energy assessment and IR spectra calculation involved performing frequency calculations using the cam B3LYP/6-311+G(d) level of the DFT method.
In addition, FT-IR calculations at the same wave number computation level were conducted. Root mean square (RMS) errors were calculated for both FT-IR methods. The optimized structure was used to calculate the geometry parameters, FMOs- frontier molecular orbitals, global electronic properties study, and molecular electrostatic potential (MEP) of the compound IQOL. For energy evaluation and IR spectral computation, FT-IR studies were undertaken using the same wave-number computation level. Furthermore, our analysis involved simulating wavenumbers in Fourier transform infrared (FT-IR).
The Gauss-Sum software program was used to generate a density of states (DOS) plot, Global electronic properties such as HOMO and LUMO, energy gap (ΔE), electronegativity (χ), and reactivity indices, including electronic chemical potential (μ), chemical hardness (η), softness (σ), ionization potential (I), electron charge transfer (ΔN), global electrophilicity (ω), and global nucleophilicity (N), were computed using the following expressions: μ = (EHOMO + ELUMO)/2, η = (ELUMO - EHOMO), ω = μ²/2η, and N = EH - EH(TCE) [40,41].
Molecular docking
Structure-based drug design often involves molecular docking analysis. In this process, the designed ligand synthesized IQOL was visualized, The energy-minimized ligand (structures) Chemoffice (Cambridge Software) in SDF formats were converted to mol2 files using open babel and the COVID-19 protein 6lu7 as downloaded from (http://www.pdb.org) website of pdb type format, further hydrogen and water molecules. The docking was conducted using the AutoDock Vina Wizard method.
The parameters for the VINA grid box size (X= -10.7290, Y = 12.4176, and Z = 68.8161) were adjusted, Use the standard exhaustiveness level of 8 to improve the binding conformation evaluation. The visual representations of entirely docked structures were created with Discovery Studio -2021 and PyMOL program package.
Pharmacological/biological assays
ADME/Tox
ADME/Tox analyses utilized both the Swiss adme and protox software of predictor. contains critical computational approaches for fully evaluating the pharmacokinetic characteristics of tiny compounds.
Results and Discussion
Chemistry
The current study, indoloquinoline alkaloid (IQOL), as shown in Scheme 1, found convincing support from 1H and 13C-NMR, FT-IR, and GC-Mass spectrums. Notably, the remaining structures only showed signals for IQOL, indicating the importance of N-H and hydroxy groups interactions of drug targets.
Synthetic routes for the desired indoloquinoline derivatives. The two-step transformation of IQOL involves an initial synthesis of 3-hydroxy-1H-indole-2-carboxylic acid 5 through two distinct routes.
Route A utilizes 1-chlorobenzoic acid 1 with glycine 2, while Route B employs ethyl bromoacetate 3 with methylantharanate 4. The subsequent step involved the reaction of 3-hydroxy-1H-indole-2-carboxylic acid 5 with aniline to produce the title compound. This procedure involves thermally induced intramolecular cyclization carried out at an elevated temperature (240 °C) under solvent-free conditions.
The suggested strategy uses the Claisen reaction technology, which involves cyclization with the help of sodium hydride. Despite the anticipation of the formation of IQOL (6), it was still present.
A possible strategy for the formation of IQOL involves employing the Claisen ester condensation mechanism, facilitated by the use of sodium hydride as a robust reagent under mediated conditions
Spectral analysis of IQOL
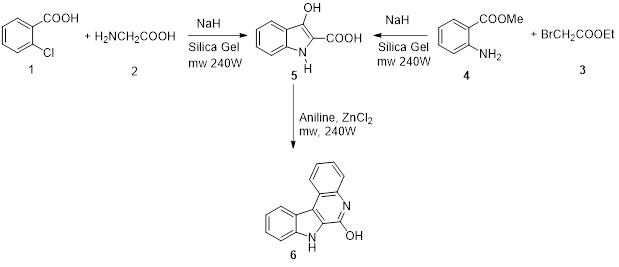
Scheme 1. Route for synthesis of indoloquinoline alkaloid analogues.
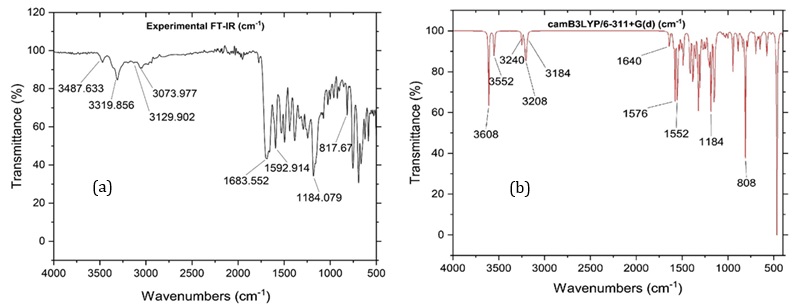
Figure 2. FT-IR spectra of IQOL (a) experimental and (b) calculated.
FT-IR spectral analysis
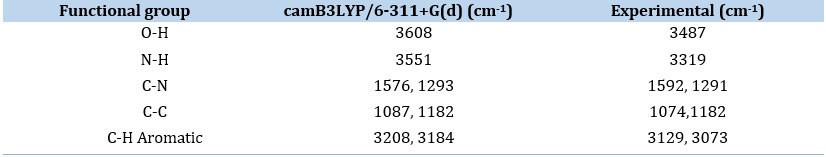
The molecular structure under discussion is an IQOL with 28 atoms, C1 point group plane symmetry, and 78 normal vibrational modes. These modes include 27 stretching, 26 bending, and 25 torsional vibrations. DFT FT-IR was obtained using B3LYP/6.311+G (d,p) parameter calculations, and then compared to experimental data. Results are shown in Table 1 and Figure 2.
CH frequency
Normally, CH stretching mode of aromatic rings is not significantly affected by substituent groups. However, specific vibrations can penetrate to substituents, providing respected structural visions toward their mechanical coupling through substituent group vibration. The 3100-3000 cm-¹ asymmetric stretching is observed. While symmetric stretches at 3000-2900 cm-¹ region.
In the ongoing analysis of the title compound IQOL, experimental observations indicated the existence of aromatic C-H asymmetric stretching 3129 cm-¹. The theoretic peak of the FTIR appeared at 3208 cm-¹. Experiments revealed symmetric v(C-H) vibrations. identified at 3073 cm-¹ with weak absorption peaks, while the calculated frequency was 3184 cm cm-¹. Comparable results were experimentally confirmed and are illustrated in Figure 2 and Table 1
C-N frequency
Assigning the stretching of C-N frequency usually found in the range of 1650-1500 cm-¹. Experimental profiles corresponding to the C-N frequency showed significant and conspicuous peaks at 1592 and 1291 cm-¹, while the computational calculations gave values of 1576 and 1293 cm-¹, respectively.
C-C frequency
The stretching vibrations of C=C bonds are spectral range of 1650-1300 cm-¹. Experimental vibrations were identified at 1496, 1385, 1291, and 933 cm-¹, with DFT computed vibration values are closely matching 1479, 1382, 1293, and 932 cm-¹, correspondingly, and experimental region of C-C stretching vibrational frequency was noted at 1074 and 1182 cm-¹, aligning well with the matching DFT computed vibrations detected at 1087 and 1184 cm-¹.
N-H frequency
Indoloquinoline ring N-H group stretching mode of frequencies, detected at 3319 cm-¹ experimentally, exhibit notable concordance with the DFT calculated slow at 3551 cm-¹.
OH frequency
The OH stretching vibrational typically ensues in the increased spectral values resulting from the electronegativity of the bond between oxygen and hydrogen atoms (OH). Affording to the literature, the reported wavenumber for the OH band is within the range of 3350-3600 cm-¹. Still, in certain instances, the OH frequency may increase after the OH group is involved in either inter-intra molecular hydrogen bonding. the OH stretching frequency was detected at 3487 cm-¹ experimentally, with a corresponding theoretical measurement at 3608 cm-¹.
NMR spectral data analysis
1H-NMR spectrum; a broad singlet for one proton is observed in the downfield region, at δ 11.52 ppm, designated as the indole-N-H proton. In addition, a broad singlet is identified in the downfield region above 15.00 ppm, assigned to the hydroxy group of the quinoline moiety.
Table 1. DFT computed and experimental FT-IR spectra of IQOL
The aromatic region displays proton signals in the downfield from δ 6.89 to 7.88, encompassing eight proton integral values corresponding to the indoloquinoline moiety, as illustrated in Figure S2.
The pertinent 13C-NMR spectrum is illustrated in Figure S3, revealing a less intense peak at 162.51 ppm, which was assigned unequivocally to the C6-OH carbon and corresponded to the hydroxy group attached to quinoline. Another less intense peak at 157.51 ppm is observed in the most downfield region, suggestive of the C7-N group carbon signal, confirming IQOL formation. This is further supported by the observation of aromatic carbon signals for (IQOL) in the range of 154.94 to 106.74 ppm.
Furthermore, the suggested structure was confirmed through the GC‒MS spectrum (Figure S4), where a (m/z) at 233.33 (observed) closely concurred with the calculated molecular mass (234). The [M-1] fragmentation peak, which accounts for 99% appeared parent peak matches perfectly the mass value of compound and is consistent with the formula for the suggested [C15H10N20]. Further support confirmed the GC-mass spectra of title compound IQOL.
DFT analysis
Optimization of IQOL
The structure of the IQOL molecule was optimized by utilizing density functional theory at the B3LYP level of computation. This approach was used to improve the geometrical characteristics of bond lengths, angles, and dihedral angles in the IQOL. The geometrical characteristics show that the indoloquinoline ring has a planar structure. The optimized geometric structure is shown in Figure 3 and Table 2.
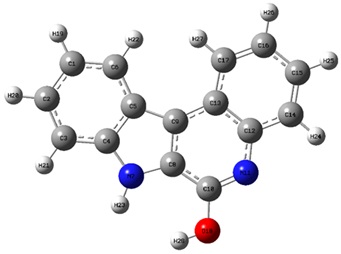
Figure 3. Optimized structure of IQOL.
Table 2. The bonds and their parameters of IQOL
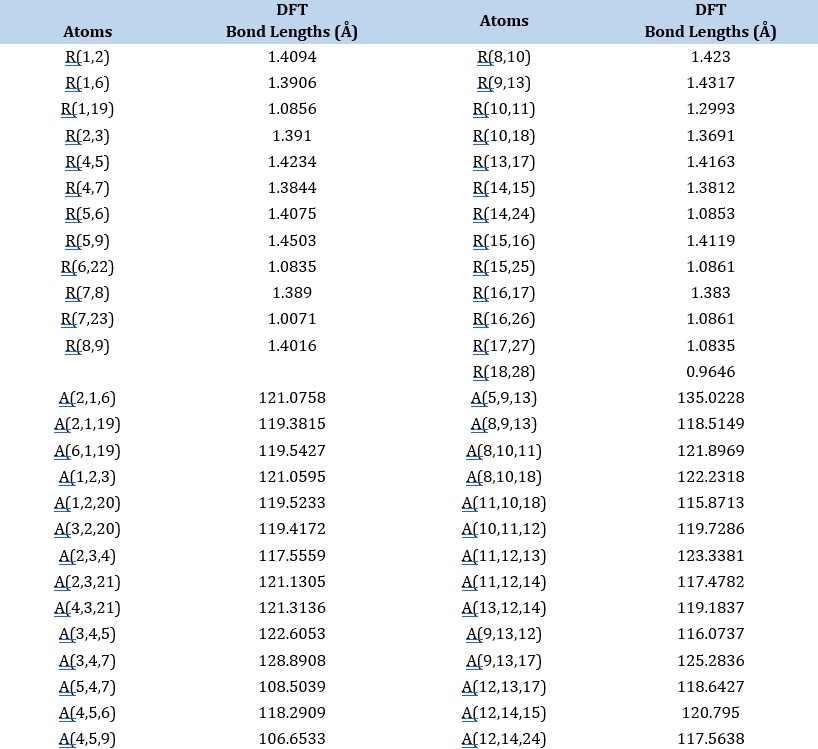

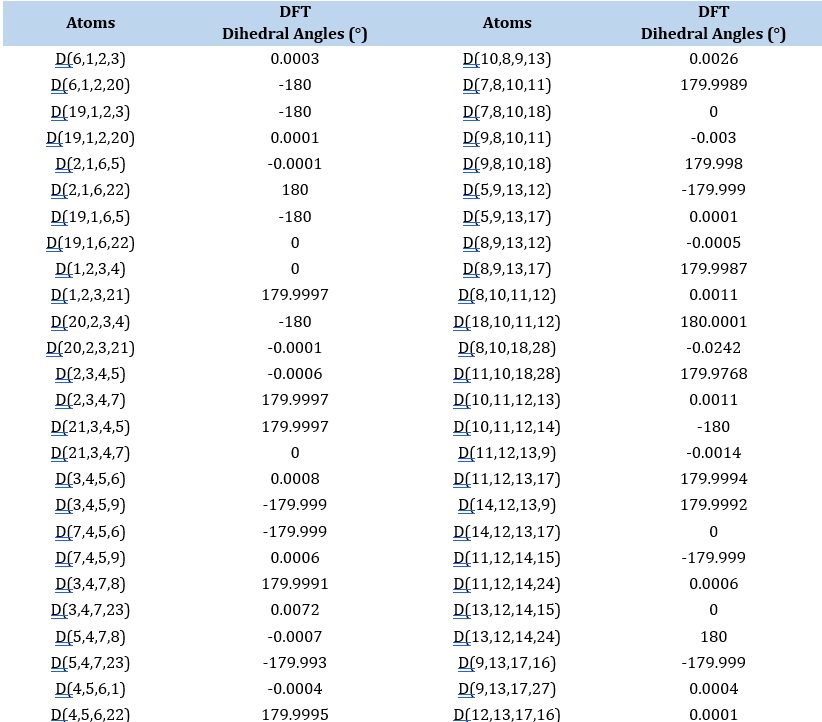
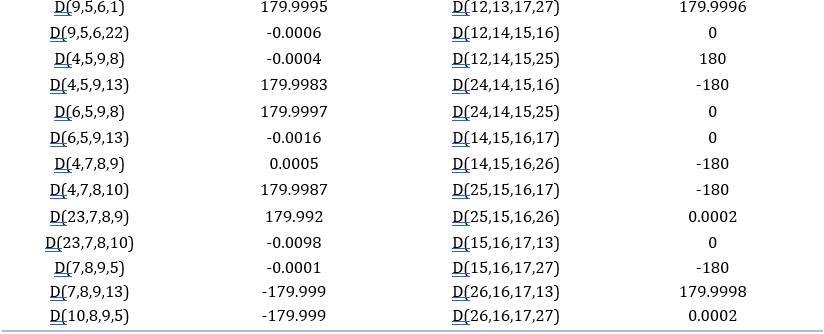
Mulliken atomic charge analysis
The analysis of Mulliken population is beneficial for approximating the charge distribution among atoms in a compound. Depending on the neighboring electrons, Mulliken charges are assigned as either positive or negative. In Figure 4 and Table 3 [40], two atoms of oxygen are negatively charged, whereas some atoms of carbon (C5, C9, C10, C12, and C13) are positively charged. In contrast, the hydroxy group connected to C possesses a negative charge. Furthermore, all atoms of hydrogen carry positive charges.
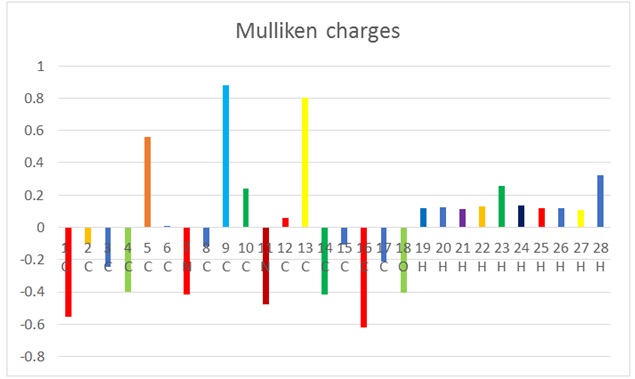
Figure 4. The Mulliken atomic charges of IQOL.
Table 3. The Mulliken atom charges of IQOL

NLO
Dipole movement, polarizabilities / hyperpolarizabilities reveal how the system reacts to a specific field approach. These parameters characterize the system's nonlinear properties, including the cross-sectional areas of different scatter along with collision operations and the power of interactions between molecules. We determined the dipole moment (μ), hyperpolarizability (β), and polarizability (α) of the IQOL compound to study the association involving the structure of molecules and nonlinear capabilities. This contact increases the molecule's polarity and is useful for intramolecular relationships. A lower band energy gap is projected to result in improved NLO properties. The hydroxyquinoline ring, functioning as an electron donor in the IQOL molecule, improves these delocalized electron densities associated with substitutes, resulting in organic NLO material properties.
The results, as outlined in Table 4, indicate values of α = 4.3313 Debye, μ = -4.276256 × 10² e.s.u., and β = 7.591340 × 10³ e.s.u. for IQOL in contrast to urea as the benchmark [42]. As a result, we deduce that IQOL has outstanding NLO (Non-linear optical) properties.
Frontier orbitals calculations (FMOs)
Molecular reactivity assessed via frontier molecular orbital (FMO) investigation, with the HOMO and LUMO playing important roles in influencing the structure's chemical stability. Eg and numerous chemical reactivity descriptors, help to characterise molecular behaviour.
Table 4. The NLO analysis of IQOL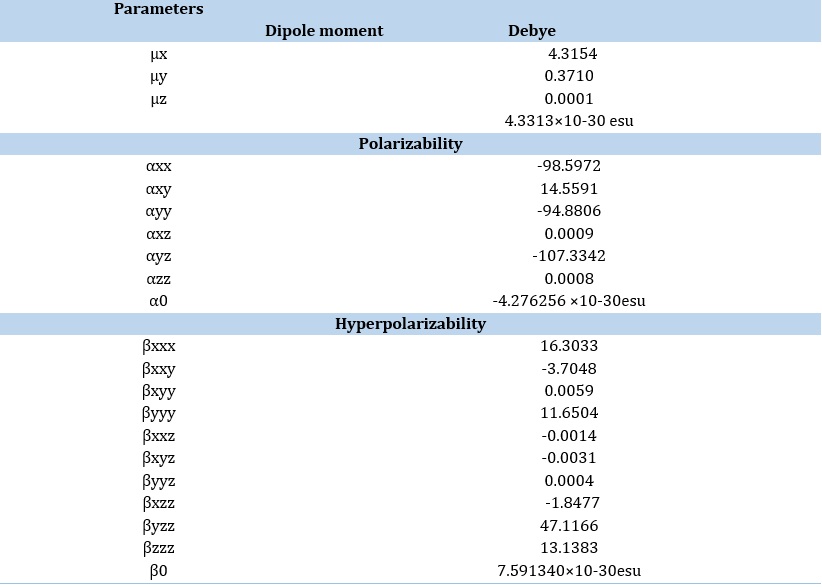
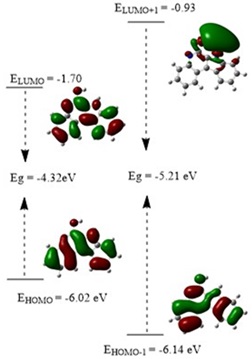
Figure 5. Energy levels of EHOMO, ELUMO, and Egap for the synthesized compound IQOL computed by the B3LYP/6-311G (d,p) method.
Figure 5 shows a graphic depiction HOMO/LUMO Properties. The global reactivity indices for IQOL were computed, as presented in Table 5. The calculated homo-lumo energy difference gap for IQOL is -4.32 eV, indicating higher chemical reactivity and a lower HOMO-LUMO energy gap. The HOMO donates electrons, whereas the LUMO accepts electrons. IQOL exhibits an electrophile nature of 3.403 and nucleophile range -4.215 eV. The evaluation of the chemical description parameter (GCRD) keys suggests that IQOL has strong electrophilic and nucleophilic properties. As expected, chemical hardness / softness is 2.284 and 1.143 eV, showing that the molecule is chemically stable and has sufficient strength.
The GCRD parameters revealed a much lower molecular value IQOL, the presence of a hydroxy group on the amine with a functional substituent. These characteristics in 3D graphs are demonstrated in Figure 5. The graphic shows that orbitals are largely found on HOMO and LUMO in indoloquinoline and its substituted systems. The Eg - gap energy, as seen in this Figure, means that the chemical effectively behaviour electrons, which favours the situation biological activity of compound.
Table 5. GCRD at IQOL in eV

DOS analysis
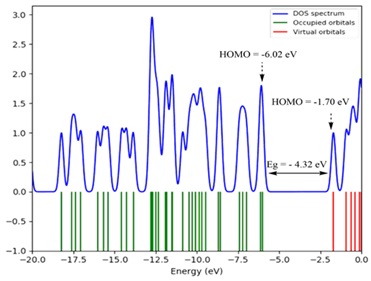
Figure 6. DOS spectrum of IQOL.
The (DOS) density of states spectrum, offers information on the amount of energy every unit of energy increment and their composition. The red and green bars on this chart show the energy values of the HOMO and LUMO, respectively. The energy of the HOMO orbital is - 6.02 eV, whereas LUMO orbital is around -1.70 eV. The energy gap Eg between the HOMO-LUMO, has been determined to - 4.32 eV. This low energy indicates allows electron movement within the indoloquinoline molecule, which is consistent with DOS spectrum measurements, as illustrated in Figure 6.
DOS intensity of 0 means that the system is not observed in any condition. The transfer of electrons between C=C and C-C in the molecule's ring creates a deviation in the peak at a given energy from its original value. The lines from −20 eV to −0 eV at the beginning of the energy axis of the Figure 6 are dubbed ''filled orbitals'', while the lines from -5 eV to 0 eV are considered ''virtual orbitals''. The vacant virtual orbitals are also known as ''acceptor orbitals'', whereas the full orbitals are known as 'donor orbitals. A high-intensity DOS at a given energy level implies that there are several states accessible for occupancy.
Molecular electrostatic potential surface
The MEP gives an extensive understanding of characteristics and interactions between intermolecular connections, making it an effective analysis for detecting electrophilic and nucleophilic locations in a molecular system. MEP emphasizes electrophilic locations, which have more attractive interactions, whereas nucleophilic locations suggest of significant repulsion. The opposing polar and nonpolar portions are graphically portrayed using different colors as in Figure. 7.
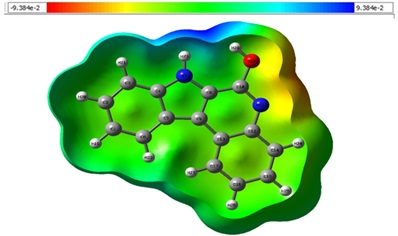
Figure 7. MEP phase for the synthesized compound IQOL.
In this representation, blue represents parts of the molecule with positive electrostatic potential values, whereas red denotes electrophilic reactivity (the most negative areas). Green and grey represent intermediate potential values between the extremes of red and dark blue. Yellow and light blue shading fill the space between the middle colour (green) and the extremes (red/dark blue). As a result, the MEP surfaces vary across the molecule from -9.384e-2 (a.u.) atomic units (shown in darkest red) to 9.384e-2 (a.u.) atomic units (shown in deepest blue). across the molecule.
Molecular docking studies
Molecular docking was a computational for evaluation the binding interactions between chemicals (ligands) and specific target proteins, notably those linked with COVID-19 key proteins such as 6LU7. The crystallographic data for COVID-19's major protease (Mpro) in association with the inhibitor N3 has been submitted in the Protein Data Bank (PDB) under the code 6LU7. This dataset provides essential insights into the complicated interplay between the SARS-CoV-2 virus's primary protease and the N3 inhibitor, as shown by X-ray crystallography. Oregon Health and Science University had a key role in developing the first COVID-19 medication, chloroquine. In molecular docking, the potential activity of the synthesised molecule IQOL was investigated by docking it into the COVID-19 site. This molecular docking technique involves testing the ligand against severe acute respiratory syndrome coronavirus 2, which is represented by PDB entries like 6LU7, to distinguish and comprehend the interactions between the ligand and the related protein.
The docking technique used the AutoDock Vina approach, which involved redocking the ligand to the active site of the target COVID-19 major protein (6LU7) for validation. The alignment of the best posture of the chloroquine reference in the active site, as well as docked structures from crystallographic ligands, indicated interactions with 6LU 7. The interactions involved four typical hydrogen bonds with the Cys145, Gln189, Thr190, and Gln166 residues, with lengths of 3.52359, 2.26388, 3.5146, and 3.49926 Å, respectively. The molecule had a Pi-Pi T-shaped hydrophobic contact with His41 (bond length: 5.49112 Å) and three hydrophobic nature pi-alkyl with His163, Pro168, and Cys145 interactions (bond lengths: 3.88 to 5.46 Å) (Figure. 8).
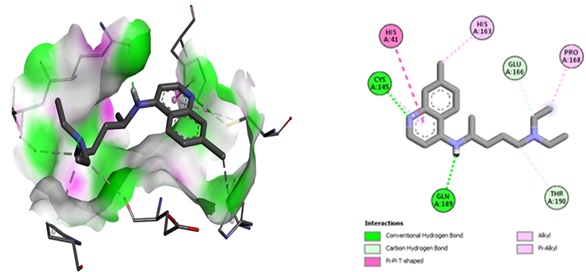
Figure 8. 2D interaction with the ligand chloroquinoline (reference): the binding site for the COVID-19 protein. PDB ID: 6LU7.
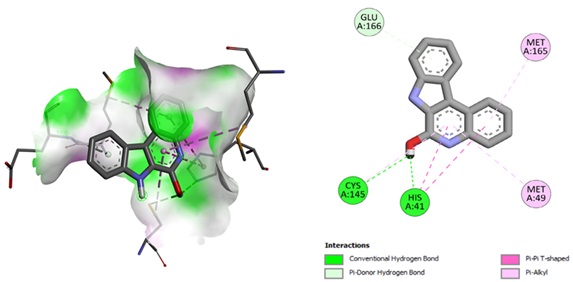
Figure 9. The docking positions of IQOL PDB ID: 6LU7.
The title compounds have low and negative binding energies, indicating a strong attraction for against COVID-19 protein. IQOL interacts with 6LU7 through three conventional hydrogen bonds interactions with Gln66, Lys145, and His41 (bond lengths: 3.898, 2.9119, 2.5666 Å), as well as two hydrophobic natures with T-shaped interactions of pi-pi bonds with His41 (bond lengths: 4.92 and 4.56 Å). There were three hydrophobic nature pi-alkyl bonds interactions found with Met165, Cys145, and Met49 (bond lengths: 4.87, 5.46, and 4.879 Å). The discovered interaction residues facilitate the complexation of the IQOL at the active of 6LU7 (Figure. 9).
Pharmacology
ADME/Tox analysis
ADME to critical pharmacokinetic characteristics used to determine pharmacological efficacy and safety. Each component of the ADME is significant for drug assessment. In the field of pharmacokinetics and drug similarity profiling, in-silico predictions are critical, as provided in Table 6. To evaluate a chemical compound's drug-like qualities, use Lipinski's rule of five (RO5), a set of criteria. These principles are often used in drug research and development to predict the likelihood of a molecule being an orally active medication in humans. Virtual screening techniques evaluate several factors. An oral drug must come across certain conditions, such as a molecular weight of less than 500 g/mol, equal to or fewer than 5 hydrogen bond donors (typically represented by OH or NH groups), equal to or fewer than 10 hydrogen bond acceptors (typically represented by O or N atoms), and a Log P (octanol-water partition coefficient) value of less than 5. It is critical to understand that following Lipinski's Rule of Five does not ensure a compounds success as a medicine; rather, it serves as a guideline for finding compounds with the potential for better oral bioavailability and a higher possibility of success in drug development. Table 6 illustrates the compound for targeting COVID-19 proteins (Figure 10).
The Tox profile, developed using artificial silico approaches, has emerged as significant analysis for anticipating the pharmacologic and toxicologic features of drug applicants, mainly in the preclinical phases (Figure 11).
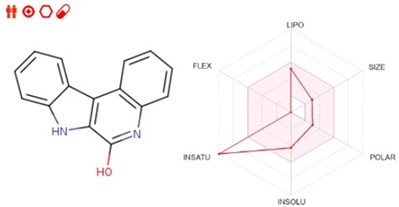
Figure 10. Bioavailability radar models of IQOL.
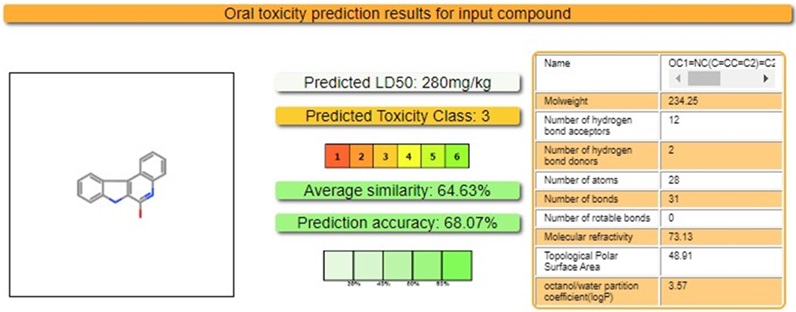
Figure 11. The in silico Tox profile of IQOL.
Table 6. ADME parametersa

BOILED-egg diagram
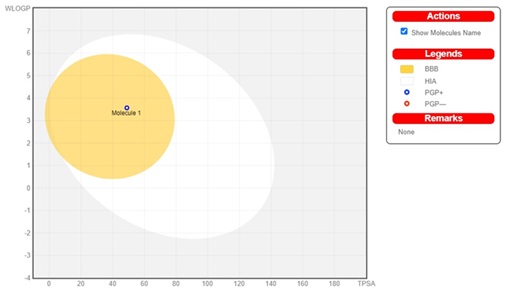
Figure 12. BOILED-Egg models of IQOL.
The study of BOILED-egg (Figure 12) shows the chemical concentration in the permitted limit for conventional medications. A bullet point with yellow region signifies that the drug remains unaffected by the CNS P-glycoprotein., whereas a blue dot in the cooked egg graphic indicates passive penetration of the blood-brain barrier. The red zone represents the capacity to persist in the brain. the synthesised ligand and its complexes satisfied the required requirements, indicating high bioavailability. These findings demonstrate that the chemical is safe and unlikely to induce skin allergies. Interestingly, the drug ratings for these substances were modest.
Conclusion
A simple and effective technique for synthesizing derivatives of IQOL, attaining exceptionally high yields via a two-step process. The synthesized compounds were thoroughly characterized using FT-IR, 1H, 13C-NMR, and GC-MS. Further travelled using DFT calculations, which revealed outstanding agreement between the expected robustness of our theoretical predictions and experimental results. Molecular docking tests demonstrated significant between the title compound and the target protein (6LU7), indicating ADME/Tox these findings highlight the possibility of these indoloquinoline (IQOL) as attractive applicants for the development of novel COVID-19.
Conflict of interest
The authors have no conflict of interest in this study.
Orcid
M. Vijayarathinam: 0009-0000-3042-2608
A. Kannan: 0009-0003-7986-4003
P. Akilan: 0009-0000-8131-3524
V. Chandrasekaran: 0000-0001-8597-9576
T. Gunasekaran: 0009-0006-5125-4918
HOW TO CITE THIS ARTICLE
Vijayarathinam, A. Kannan, P. Akilan, V. Chandrasekaran, T. Gunasekaran. A Novel Approach in the Synthesis of Indoloquinoline Alkaloid Analogues: Spectroscopic and DFT Exploration, Molecular Docking of COVID-19, and ADMET Properties. Adv. J. Chem. A, 2024, 7(4), 417-437.
DOI: 10.48309/AJCA.2024.437859.1484
OPEN ACCESS
©2024 The author(s). This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit: http://creativecommons.org/licenses/by/4.0/
PUBLISHER NOTE
Sami Publishing Company remains neutral concerning jurisdictional claims in published maps and institutional affiliations.
CURRENT PUBLISHER
Sami Publishing Company
)