Document Type : Original Research Article
Authors
Department of Chemistry, Faculty of Physical Science, Ahmadu Bello University, P.M.B. 1044, Zaria, Kaduna State, Nigeria
Abstract
Hepatitis C virus (HCV) infection promotes death rates worldwide. As a result, there is a constant need to improve current HCV therapy and produce new drugs. The NS3/A4 enzyme plays a critical role in the HCV entire lifespan and proliferation. Consequently, inhibitors of the HCV NS3/A4 enzyme are a great spot to start exploring new drug candidates. In this study, the high throughput in silico screening of the Pubchem database was used to analyze a set of ketoamides as HCV NS3/A4 enzyme inhibitors to find a novel potential drug as a lead candidate. To Voxilaprevir as a reference medicine, our findings revealed that three HCV NS3/A4 protease inhibitors (Pubchem CID: 44158040, 44158107, and 11479303) were identified as the best drugs for blocking hepatitis C virus NS3/A4 protease. The QSAR was performed to study the relationships between the structural features of the targets and their binding affinity by developing statistical models. The reported compounds had a higher binding affinity for the target receptor than Voxilaprevir, the reference drug. This study could be important in understanding the physicochemical and binding affinity of HCV NS3/A4 inhibitors in order to find new and improved HCV antiviral drugs.
Graphical Abstract
)
Keywords
Introduction
Hepatitis C virus (HCV) infection promotes death rates worldwide. Persistent HCV infection is a serious health problem worldwide, impacting up to 3% of the populace and killing over 300,000 people annually [1-3]. Its gradual spread and hard-to-detection make it a hidden pandemic, and most infections proceed into a persistent condition that lasts for years. About 60%-80% of people infected with HCV suffer persistent hepatitis, with 20% developing cirrhosis and about 2%-5% of patients dying from liver cirrhosis and malignancy [4,5].
In 2019, there were approximately 58 million severe recorded cases around the globe, with 75% of them occurring in low- and middle-income countries (LMICs) [6]. New HCV infections are occurring in high-risk populaces worldwide, including drug users and men who have sex with males [7]. HCV is primarily transmitted in Sub-Saharan Africa through improper medical practices and infected blood transfusions. Needlestick injuries in medical professionals, mother-to-child spread (MTCT), and societal behaviors like piercing and tattooing are also possible transmission routes [7,8].
The hepatitis C virus gene contains a polyprotein with around 3000 amino acids that are separated into two groups: The structural and non-structural (NS) proteins are the critical targets for antiviral drug development because of their critical role in HCV virus propagation [2,9]
Current HCV treatment has a poor sustained viral chance of success, the rapid development of drug resistance, and significant side effects, leading to treatment discontinuation [10]. As a response, more effective anti-HCV drugs having minimal adverse effects are desperately required. The discovery of the most critical information to facilitate the identification of unique bioactive compounds that could be utilized to cure HCV infection and then provide medically successful therapy is undoubtedly a critical global health priority [11].
Pharmaceuticals can be designed in silico using molecular docking by enhancing lead candidates targeted against specific receptors. A docking method can be used to find the best small molecule (ligand) binding mode to an active receptor domain. As a result, drug research aims to generate molecules that are bound to a receptor more strongly than the native ligand [12]. The biological reaction catalyzed by the target molecule can be modified or inhibited in this way. Cheminformatics has become extensively utilized in pharmaceutical research due to the many advantages of being quite time-consuming, cost-effective, and high reliability in the computational screening and modeling of viable drugs. High-throughput screening techniques that employ in vitro tests to determine the biological responses of a variety of molecules towards a specific macromolecule are often used to identify drugs by chance in a trial-and-error procedure. This is a lengthy and expensive process. If the target's 3D structure is known, a program to mimic docking can help drug discovery. This in silico study identifies promising therapeutic candidates more quickly and at a lower cost by virtual screening of drug repositories.
Laboratory investigations (synthesis), cytotoxic screening, clinical trials, and other procedures could be utilized to investigate the drug molecules discovered using the in silico approach. Most docking approaches employ an energy-based ranking system to find optimum thermodynamically stable ligand orientation when a ligand is attached to a target. Lower energy scores are assumed to indicate better protein-ligand bindings when compared to higher energy scores. Consequently, docking can be conceived as an iterative algorithm aiming to find the ligand-binding formation with the least energy [13]. The present research seeks to discover possible pan-genotypic HCV NS3/4A protease inhibitors with enhanced pharmacokinetic profiles and lower hepatotoxicity threats. In this article, we used a docking study to screen the Pubchem database for possible lead (hit) molecules. We further build a QSAR model to find underlying molecular characteristics that can facilitate the discovery of new biomolecules as anti-HCV medications.
Materials and Methods
Dataset
53 ketoamides compounds were used as possible inhibitors of the HCV NS3/4A enzyme, which were retrieved from a website (https://pubchem.ncbi.nlm.nih.gov/). Table SM1 in the Supplementary Material contains the datasets and Pubchem compound identification number (CID) and Energy score (Escore).
Optimization
Optimizing a molecule's equilibrium or concept energy geometry is referred to as optimization. Chemdraw v 12.0 was used to sketch the structures of the molecules, which was then transferred to the Spartan 14 program to minimize the geometry using B 3 LYP level of theories and 6-31G as the basis set [13].
Target preparation and docking procedure
The receptor utilized in this research was retrieved from a web database (PDB) uploaded on 14th February 2019 by Taylor, and a co-worker [14] with the relevant details: PDB ID (6NZT), the resolution (1.40 Å), R-value free (0.197), the R-value work (0.176), and the R-value observed (0.177). The native molecule was detached from the receptor before being introduced into the Pyrx software for docking. As discussed in our prior research, the comprehensive docking strategy was applied [15]. Figure 1 illustrates the receptor.
Theoretical background of the energy terms in the docking Algorithm
The energy terms that make up the binding energy scoring function, Escore, are presented in Equation 1. ![]()
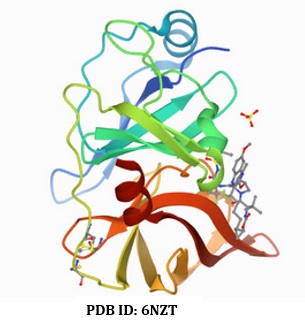 Figure 1. Structure of the receptor (HCV NS3/A4 protease) in complex with native ligand (Voxilaprevir)
Figure 1. Structure of the receptor (HCV NS3/A4 protease) in complex with native ligand (Voxilaprevir)
Einter represents protein-ligand interaction energy, shown in Equation 2.
![]()
All heavy atoms in the ligand and protein, together with any coenzyme atoms, may be present. The EPLP word refers to a piecewise linear potential, further explained down. The second phrase describes attractive forces between charged atoms. It is a Coulomb potential with the dielectric constant D(r)=4r, which varies with distance. The last term in Equation 2 gives the clash penalty of the electrostatic energy in kcal/mol and a threshold value with a distance of 2.0 Å, so that distances < 2.0 Å confirm that no energy input is greater than the clash penalty.
Eintra is the internally potential of the ligand, given by Equation 3.
![]()
The double summation applies to all-atom sets in the ligand, omitting those bound by two or fewer bonds. The second part is a torsional energy factor parameterized by the bonded atoms' hybridization kinds. represents the bond's torsional angle in degrees. If many torsions could be established, the mean of the torsional energy bond input was employed. If the difference across two heavy atoms (more than two bonds apart) < 2.0 Å, the last part, Eclash, imposes a penalty of 1000. As a result, the Eclash term penalizes impracticable ligand conformations.
QSAR method
The datasets were split into model building data and model validation data of 70% and 30%, respectively, as outlined in our prior study [2].
Accessing the identified entities
The web applications SwissADME (www.swissadme.ch/) and pkCSM (http://structure.bioc.cam.ac.uk/pkcsm) were used to quantify the drug-likeness and pharmacokinetics ADMET profile of these newly discovered entities to verify their reliability.
Result and Discussions
In this study, the most thermodynamically viable ligand geometry was identified, and lead (hit) molecules were discovered as HCV NS3/4A enzyme inhibitors using computational docking models, with docking scores ration as binding energy. The majority of the screened molecules are potent against the HCV NS3/4A enzyme, and the best HCV NS3/4A enzyme inhibitors were ranked according to their Lower energy docking score. The docking results revealed that molecules 6, 15, and 45 were the best hits because they established covalent and noncovalent interactions at the receptor's binding domain. Also evidenced by the energy terms and their binding sequences (Figures 2, 3, and 4, respectively) seem to be consistent with that of the standard drugs (Voxilaprevir) (Figure 5). Those molecules pose maximum interactions with the critical amino acid residues in the target binding domain as detailed in Tables SM2, SM3, SM4, SM5, respectively.
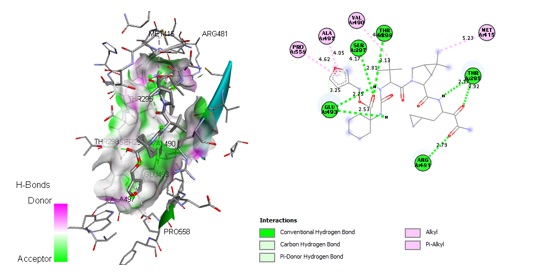 Figure 2. The 3D and 2D representation of compound 6 (CID: 44158040) with the target receptor (PDB ID: 6NZT)
Figure 2. The 3D and 2D representation of compound 6 (CID: 44158040) with the target receptor (PDB ID: 6NZT)
 Figure 3. The 3D and 2D representation of compound 15 (CID: 44158107) with the target receptor (PDB ID: 6NZT)
Figure 3. The 3D and 2D representation of compound 15 (CID: 44158107) with the target receptor (PDB ID: 6NZT)
 Figure 4. The 3D and 2D representation of compound 45 (CID: 11479303) with the target receptor (PDB ID: 6NZT)
Figure 4. The 3D and 2D representation of compound 45 (CID: 11479303) with the target receptor (PDB ID: 6NZT)

Figure 5. The 3D and 2D representation of reference compound (R) with the target receptor (PDB ID: 6NZT)
The identified molecules were then evaluated for drug-likeness using the Lipinski concept of five, which essentially asserts that if a compound infringes over two of the characteristics listed in Table 1, it will be improperly absorbed [15] and is one of the most valuable approaches in the initial stages of drug improvement. Because the discovered molecules did not break more than two of Lipinski's threshold, they are likely to be classified as drug-like molecules. Additionally, ABS standards were used to evaluate the template and all discovered compounds, including the standard molecule, with values of 0.17 (17%). The reliability coefficient of a molecule with excellent permeability and bioavailability pattern reflects total compliance with the Lipinski idea of five, which gives a value of more than 10%, is reliable [16]. The synthetic accessibility of the compounds in consideration was also assessed on a scale of 1 to 10 (very simple to synthesize to difficult to synthesize) [16]. As shown in Table 1, the discovered compounds have synthetic accessibility ranging from 6.02 to 6.38, making them moderately easy to synthesize.
Table 1. Derived drug-likeness focusing on Lipinski's rule

Using web-based pkCSM programs, the ADMET profiles of discovered molecules and the standard molecule have also been analyzed and listed in Table 2. The BBB penetration ratings are used to establish if a substance will pass through the blood-brain barrier (BBB). A Log BB > 0.3 suggests that a molecule can readily flow through the blood-brain barrier, whereas a Log BB < -1 suggests that such a molecule was poorly dispersed [15]. In this study, all of the molecules investigated efficiently penetrated the blood-brain barrier (Table 2).
A compound with a Papp > is reported to have the highest Caco-2 permeability, which can be used to estimate orally administered permeability in vitro [15]. According to the findings of this investigation, all of the molecules investigated have only a moderate cell uptake in Caco-2 cells.
The HIA estimates how much of a chemical will be consumed via the human gut. Compounds with an absorption rate<30% are considered poorly consumed [17]. The investigation results demonstrated that all of the substances investigated have high HIA ratings.
The maximum tolerated dose (MRDT) is a metric for determining a chemical's toxic dosage limit in humans. A low MRTD for a molecule is less than or equal to 0.477 log(mg/kg/day), whereas a high MRTD is greater than 0.477 log(mg/kg/day) [15,16]. Table 2 reveals that the lethality of all of the chemicals studied in this study was minimal.
Table 2. ADMET profiles of discovered molecules, and reference molecule

Equating the binding affinities of the molecules with their structural features and constructing comprehensive statistical models using both linear (multiple linear regression and partial least squares regression) and non-linear (support vector machine, and neural network regression) simulation techniques which give an insight into the nature of the interaction and the model has been proposed using the equation below:
![]() (4)
(4)
The model fulfilled the requirements specified in Table 3 as a baseline, indicating that it can make accurate predictions. The model also fulfilled Tropsha and the OECD criteria [12,18] as it explains above 90% and predicts above 70% of the variations of the HCV NS3/4A protease inhibitors with their binding affinity using both linear and non-linear simulation techniques as described in Table 3. It shows that the model accurately extrapolated the data and can estimate its appropriate building data. It estimated more than 70% of the data and thus fulfilled the requirement of 60% [12].
Table 3. Model external validation parameters and their threshold values using linear (MLR and PLS) and non-linear (SVM and ANN) methods

The meaning and nature of the descriptors were presented in supplementary material Table SM6 with the descriptor’s variance inflation factor (VIF) for better understanding. Also, the variance inflation factor (VIF) is represented pictorially in Figure 6, demonstrating that all the descriptors have VIF coefficients lesser than 5. Therefore, the developed model is statistically significant, and the descriptors are reasonably orthogonal. Figures 7, 8, 9, and 10 reveal the estimated vs. actual binding affinity throughout the original data for Multiple Linear Regression, Partial Least Squares Regression, Support Vector Machine, and Neural Network Regression, respectively. The Figures revealed a close relationship between actual and predicted binding affinity based on the linearity of these graphs.
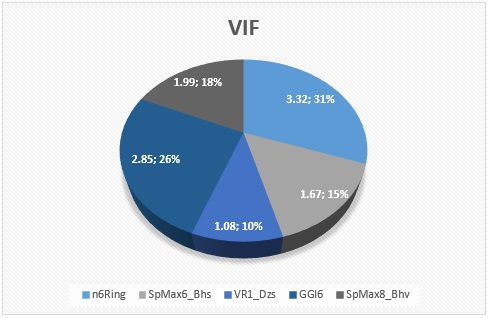
Figure 6. Chart describing the variance inflation factor (VIF) of the model
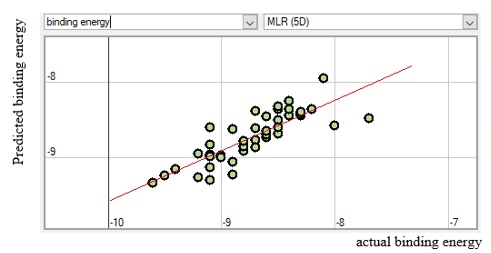
Figure 7. MLR Predicted binding energy against actual binding energy

Figure 8. PLS Predicted binding energy against actual binding energy
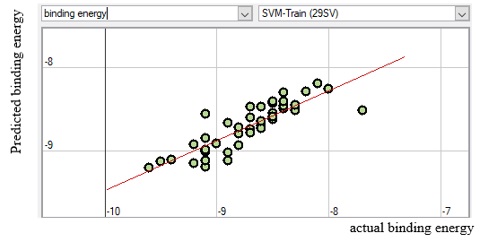
Figure 9. SVM predicted binding energy against actual binding energy
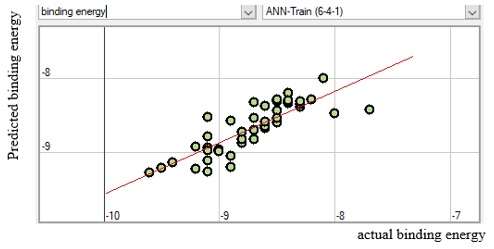
Figure 10. ANN Predicted binding energy against actual binding energy
Conclusions
Our findings revealed three HCV NS3/A4 protease inhibitors (Pubchem CID: 44158040, 44158107, and 11479303) were the best drugs for blocking the HCV NS3/A4 enzyme in comparison to Voxilaprevir as a reference medicine. By establishing statistical models, the QSAR was determined to investigate the correlations between the molecular structures of the targets and their binding affinity. The identified molecules exhibited better binding affinity than Voxilaprevir, the reference drug, with the target receptor. ADMET estimate revealed improved druggability with lower MRTD of those identified molecules. Based on the molecular docking, QSAR model, drug-likeness, and ADMET analysis, this investigation could be useful to understand the structural profile and binding affinity of HCV NS3/A4 protease inhibitors to search for novel and better HCV antiviral drugs.
Acknowledgment
Authors are grateful to the Physical chemistry research team head by Prof. Uzairu of department of the chemistry Ahmadu Bello University Zaria-Nigeria for their meaningful contributions.
Disclosure statement
The authors reported no potential conflict of interest.
ORCID
Stephen Ejeh : 0000-0002-3065-6575
)