Document Type : Original Research Article
Authors
1 Department of Chemistry and Industrial Chemistry, Kwara State University, Malete, Ilorin, Nigeria
2 H.E.J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, Pakistan
3 Dr. Panjwani Center for Molecular Medicine and Drug Research, International Centre for Chemical and Biological Sciences, University of Karachi, Karachi 75270, Pakistan
Abstract
Obesity, a lipid metabolic disorder characterized by excess fat deposition in the adipose tissue, is among the leading top global health challenges. The only Food and Drug Agency (FDA) approved drug (Orlistat®) for its treatment has shown some adverse effects. To find new compounds that may be more effective or with less adverse effects compared to Orlistat®. Catechin and chlorogenic acid were computationally studied using molecular docking and validated with molecular dynamics simulation techniques. The ADMET and drug-likeliness evaluation of the two compounds was carried out in silico. The binding affinities, structural stability, and flexibility vis-a-vis root-mean-square deviation (RMSD) and root-mean-square fluctuations (RMSF) plots, hydrogen bonding, and surface area analysis of the two compounds were compared to the Orlistat®. It was found that the selected two compounds passed Lipinski’s rule of 5 and other parameters expected of a drug. In addition, both catechin and chlorogenic acid exhibited good docking scores, better fit and molecular interactions, good structural stability, and flexibility compared to Orlistat®.
Graphical Abstract
)
Keywords
Introduction
According to the world health organization (WHO), recent worldwide reported health cases of obesity have nearly tripled those of 1975 with over 650 million obese populations [1]. Governments and health agencies in the developed and developing countries are highly concerned about obesity due to the multitude of problems such as hypertension, hyperlipidemia, diabetes mellitus, cardiovascular disease, cancer, and metabolic disorders [2–4]. In the cases where diets and increased physical activity fail to show positive results, pharmaceuticals, such as Orlistat®, an analog of lipstatin obtained from Streptomyces toxitricini, are recommended [5, 6]. Orlistat®, a pancreatic lipase inhibitor, is the only OTC (over-the-counter) anti-obesity drug approved by the food and drug agency (FDA) for the long-term treatment of obesity. However, recent findings reported severe adverse effects such as liquid stool, incontinence, flatulence, and abdominal colic, with its long-term administration [7, 8]. Therefore, there is a need to explore and discover more effective, cheaper, and better alternatives with lesser side effects.
Several works have been conducted on exploring plant-based natural products, which represent a vast reservoir of chemical entities that have the potential to treat various metabolic disorders.
From the literature survey, many natural products (plant extracts and isolated compounds) have been reported for their pancreatic lipase inhibition property, including catechin and chlorogenic acid [9]. Sahib et al., [2] evaluated the in vitro antipancreatic lipase activity of ethanolic extract of Centella asiatica, Morinda citrifolia, and Momordica charantia (fruits) at various concentrations (7.81–250 ppm) using Orlistat® and catechin as synthetic and natural positive controls, respectively. The plant extracts Morinda citrifolia, Momordica charantia, and Centella asiatica fruits inhibited pancreatic lipase activity with 21.0±1.3, 25.8±0.1, and 25.3±0.4% inhibition, respectively. Cho et al., [10] investigated the efficacy of chlorogenic acid on altering body fat in high-fat diet (37% calories from fat) induced-obese mice compared to caffeic acid and reported that chlorogenic acid seemed to be more potent for body weight reduction and regulation of lipid metabolism than Caffeic acid. Kumar et al., [11] explored the medicinal promises of chlorogenic acid, emphasized its antiobese property, and revealed that chlorogenic acid shows promise as an antioxidant, glycemic control agent, anti-hypertensive, anti-inflammatory, antimicrobial; neuroprotective, and anti-obesity agent. Saeki et al., [12] discussed the catechin-protein interaction as part of the mechanism of catechin’s beneficial actions in health. They noted the importance of computational docking analysis (CMDA) and X-ray crystallographic analysis (XCA) in elucidating the action and mechanism of catechin-protein interactions. Zhou et al., [13] evaluate the inhibitory effects of different phenolic extracts from non- and ultra-high pressure- (UHP)-treated palm fruits and their main phenolic compounds against pancreatic lipase and α-glucosidase and to further analyze the interaction and inhibitory mechanisms of two primary phenolic (caffeic acid and catechin). Results showed that the free, esterified, and insoluble-bound phenolic fractions from the non- and UHP-treated fruits demonstrated good inhibitory effects towards two enzymes. Mutya and Paul [14] performed molecular docking and experimental validation of some pancreatic lipase inhibitors to gain insight into the molecular interaction between the ligand and the enzymes. Recently, many researchers have adopted computational methods such as molecular docking, virtual screening, and qualitative-structural activity relationship to study the effects of drugs and vitamins on the human body [15–19]. Though molecular docking is good at revealing the best fit and pose, it is limited as a technique to provide insight into stable molecular interactions between docked protein-ligand complexes under specific physiological conditions. In this study, the toxicity, drug-likeliness, and inhibitive property of catechin and chlorogenic acid against human pancreatic lipase were investigated computationally through ADMET studies, molecular docking, and molecular dynamics simulation methods to determine their suitability as better alternatives to Orlistat®.
Computational Methodology
Ligands preparation
The molecular structures of the ligands (catechin, chlorogenic acid, and Orlistat®) obtained from PubChem’s structural database [20] were converted from SDF format into PDB format using Open Babel GUI 3.11 [21]. Using Merck Molecular Force Field (MMFF) method and conformation search methods, a reasonable input geometry with minimized energy of stable conformation was generated using the Spartan 14® 1.1.4 version [22]. The resulting conformers were optimized using the equilibrium geometry Density Functional Theory (DFT) method at B3LYP and 6-31+G* basis set per Equation 1.
![]() (1)
(1)
Where E is the potential energy of reasonable or stable geometry, Estretch is the stretching vibrational energy; Ebend is the energy associated with bond angles due to elastic bending for optimization; Etorsion is the energy associated with torsional energy, and Enon-bonding is the energy that keeps the non-bonding electrons in their orbital space.
Protein preparation
The target receptor, the crystal structure of the human pancreatic lipase (HPL) complexed with cofactor, procolipase with PDB code: 1N8S [23] was obtained from the Protein Data Bank (www.rcsb.org) webserver [24]. BIOVIA Discovery Studio Visualizer® 2021 version [25] was employed to remodel the missing residues on chain A (i.e., residues 501-505 of lipase) and chain C (i.e., residues 591-595 of colipase). Since the complex contained neither water molecules nor any experimental inhibitor, polar hydrogens were modeled directly using AutoDock Tools® 1.5.6 version [26], and Gasteiger charges were computed to neutralize the complex. All the amino acid residues in the active site of the HPL-colipase complex though earlier reported in the literature [27–29] were confirmed computationally using the Computed Atlas for Surface Topography of proteins (CASTp) webserver [30].
ADMET and drug-likeness studies
To predict the toxicity level of catechin and chlorogenic acid, in-silico toxicity studies were performed using the admetSAR 2.0 webserver (lmmd.ecust.edu.cn/admetsar2/) [31] and the SwissADME® tool, where the bio-availability radar was obtained [32]. The drug-induced hERG toxicity, carcinogenicity, human oral bio-availability, AMES mutagenesis, acute oral toxicity, water solubility bio-degradation, and other parameters were computed from the server.
Molecular docking and molecular dynamic simulation studies
All the ligands and macromolecules in PDB format were saved as (PDBQT) using AutoDock Tools® 1.5.6 version. AutoDock/Vina Tool® 1.5.6 version [33] was applied primarily for docking and calculating the binding energies of ligands’ respective poses. The grid box (X=40.552, Y= 18.551, and Z= 84.308) was set in response to the active sites determined. The docking results revealed the binding affinity/ligand efficiency (∆G) and the inhibition constant (Ki) of the three ligands (Table 1).
Molecular dynamics simulation studies were carried out by employing the NAMD 2.0 version [34] with the minimum energy configurations of the compounds using CHARMM force field. For ligands parameterizations and topology, CHARMM GUI webserver (www.charmm-gui.org/) [35] was used. Solvation of the system was done via simple point charge model (SPC/E) water in a cubic box, leaving 5.0 nm space around the solute. Counter ions in the form of Na+ of concentration, 0.15 M ions were added to neutralize all the systems.
Energy minimization of the system was carried out to reach a maximum force. The system was then equilibrated for 10000 ps at 300 K using an Isochoric-Isothermal or (NVT) ensemble followed by Isothermal–Isobaric or (NPT) ensemble for another 10000 ps of equilibration at 300 K. For both NVT and NPT, equilibrations, the electrostatic and van der Waals interaction cutoffs were fixed at 1.0 nm. These equilibrated ensembles were then subjected to Molecular Dynamic simulation for 100 ns with the same electrostatic and van der Waals cutoffs.
The trajectories and structures of the complexes were observed and visualized by VMD (Visual Molecular Dynamics) [36]. For graphical analysis, the Xmgrace® tool [37] was utilized to plot the graphs of Root Mean Square Deviation and Fluctuation (i.e., RMSD and RMSF), hydrogen bonding, Radius of Gyration (RoG), and Solvent Accessible Surface Area (SASA).
Results and Discussion
Binding affinities and inhibition constants
Table 1 shows catechin and chlorogenic acid's binding affinity or inhibitory properties against the human pancreatic lipase receptor with Orlistat® as standard. Since binding affinity is a function of the inhibitory properties of selected inhibitors, it is clear that catechin and chlorogenic acid has a better binding affinity to the human pancreatic lipase crystal complex than Orlistat®, the control drug.
Steven (2013) [38] submitted that the expected inhibition constants for any potential drug in drug design processes should be in the range of 0.01 µm - 100 µm. The inhibition constants for the two selected drug candidates (i.e., catechin and chlorogenic acids) against the human pancreatic lipase fall within 0.08 µM and 0.18 µM (Table 1). The graphical representation of the docking results (using the binding affinity) is presented in Figure 1.
Table 1. Chemical descriptors (molecular weight, binding affinities, and the inhibition constants) of the three ligands against the human pancreatic lipase crystal structure

 Figure 1. The graphical representation of the docking results for chlorogenic acid, catechin, and Orlistat®
Figure 1. The graphical representation of the docking results for chlorogenic acid, catechin, and Orlistat®
Binding modes and binding interactions
Tables 2 and 3 demonstrate the binding modes and interactions of catechin, chlorogenic acid, and Orlistat® with the human pancreatic lipase enzyme. From the results, catechin formed 9 interactions: 2 strong hydrogen bonds and 7 weak electrostatic interactions with the binding sites of the receptor. Likewise, chlorogenic acid forms 9 interactions with 6 strong hydrogen bonds and 3 weak electrostatic interactions with the receptor. On the other hand, the control drug, Orlistat® forms 6 interactions with 3 strong hydrogen bonds and 3 weak electrostatic interactions within the receptor’s binding sites. It is convenient to infer that catechin and chlorogenic acid cling better to the cavity of the binding pockets of human pancreatic lipase than the control drug, Orlistat®.
The pictorial representation of the interactions of the ligands with the receptor are shown in Figures 2a-2c.
Table 2. Binding sites interactions and binding distances between the ligands and the protein residues

Table 3. Binding interactions of Catechin, Chlorogenic acid and Orlistat with the protein residues


 Figure 2a. Catechin at the human pancreatic lipase binding pocket
Figure 2a. Catechin at the human pancreatic lipase binding pocket
 Figure 2b. Chlorogenic acid at the binding pocket of the human pancreatic lipase receptor
Figure 2b. Chlorogenic acid at the binding pocket of the human pancreatic lipase receptor
 Figure 2c. Orlistat® at the binding pocket of the human pancreatic lipase receptor
Figure 2c. Orlistat® at the binding pocket of the human pancreatic lipase receptor
ADMET and drug-likeliness studies
Table 4 shows that catechin and chlorogenic acid are water-soluble compared to the solubility of Orlistat® (standard), which is one of the critical conditions, a drug must satisfy before in vitro drug optimization process. Cytochrome CYP450 inhibitors are responsible for the biotransformation of a drug, and a potential drug should not alter the kinetics. Results showed that catechin and chlorogenic acid are non-inhibitors of cytochrome CYP450 inhibitors (with 0/5 rating for both compounds). In contrast, Orlistat® (standard) is predicted to be an inhibitor of at least one of the CYP450 inhibitors.
The brain blood barrier (BBB) prediction test suggests that catechin and chlorogenic acid cannot cross the BBB. Therefore, their mode of action does not require neurological engineering, such as appetite reduction, as observed in Orlistat® (standard) in the treatment of obesity. It also suggests that they will pose little or no side effects on long-term administration compared to Orlistat®, which was reported to cause intense side effects in long-term use [39].
The human either-a-go-go related inhibition measures the electrical pulse transmission in the human heart (iodine-channel blocking). A potential drug should not block the activity of the hERG inhibitors [40]. The predicted result indicated that catechin, chlorogenic acid, and Orlistat® (standard) will not result in hERG inhibition.
AMES toxicity measures the potential for DNA mutation. Therefore, a potential drug should not be AMES toxic [40]. The AMES toxicity prediction test revealed that catechin and chlorogenic acid are non-AMES toxic.
Table 4. ADMET parameters computed from admetSAR 2.0 web servers and SwissADME® tool for predicting the drug properties of catechin, chlorogenic acid, and Orlistat®.

The thyroid hormonal gland is a hormone synthesized and secreted by the adenohypophysis, which stimulates the thyroid gland to produce the hormones thyroxin and tri-iodothyroxine. Thyroxin is an iodine-containing poly-peptide hormone secreted by the thyroid gland and is essential for normal cell metabolism and several other body developments. Therefore, a promising drug should not alter its natural functions. Results demonstrated that the chlorogenic acid would not bind to the thyroid receptor sites, but catechin would. However, the value between the accepted value and catechin’s binding possibility is negligible when subjected to further drug optimization.
Estrogen receptors are naturally occurring steroid compounds formed in the ovary. They can also be synthetically prepared. Estrogens have hormonal activities which are essential for normal female sexual development. Among the more important are estrogen and estradiol. A potential drug should also not alter its natural function. These results suggest that catechin will not bind to the estrogen receptor, but chlorogenic acid and Orlistat® would. This factor (Estrogen receptors) is optimizable during the further drug preparation process.
Drug-Likeliness
Table 5 shows that catechin and chlorogenic acid do not contain heavy atoms that can be difficult to breakdown (bio-transformation) compared to the standard drug, Orlistat®. Both catechin and chlorogenic acid passed Lipinski’s rule of 5 [41] regardless of chlorogenic acid defaulting with its number of ionizable hydrogen ions (i.e., 1/5), which can be optimized in further steps case of the standard drug, Orlistat®. Catechin and chlorogenic acid also have more potential to behave as a buffer considering their hydrogen bond donor and accepting abilities than Orlistat®.
Table 5. Drug likeliness of catechin, chlorogenic acid and Orlistat®

Root-Mean-Square Deviation (RMSD)
The RMSD is used to measure the structural differences between structures and complexes, i.e., to estimate changes in the molecular conformations. In Figure 3, the RMSD plot indicates the initial fluctuations for all complexes expected due to the solvation in the water box, which restores the dynamic motions of all complexes. From the graph, the catechin complex (black color) experienced initial fluctuations up to 5 ns. It became stable throughout the whole simulation time with an average RMSD value of 0.26 Å, followed by chlorogenic acid complex (red color) with fluctuations up to 40ns (RMSD value of 0.22 Å) and Orlistat® complex (green color) shows fluctuations up to 55ns after which its RMSD values (0.42 Å) become constant during the simulation time and specified unimportant changes in the atomic fluctuations of all the structures. Stone et al., [42] submitted that for any given RMSD trajectory calculation, a backbone RMSD around 0.2 nm (2 Å) is a pointer to small structural fluctuation. In comparison, a structural fluctuation greater than 0.3 nm (3.0 Å) indicates a substantial conformation structural change. Since the time required for the simulation system to reach equilibrium for chlorogenic acid is around 40 ns and for catechin is 5 ns, it can be inferred that catechin and chlorogenic acid complexes exhibit better conformational changes than Orlistat® complex throughout the simulation.
 Figure 3. The backbone RMSD plot of chlorogenic acid (red), Orlistat (green), and catechin (black) complexes
Figure 3. The backbone RMSD plot of chlorogenic acid (red), Orlistat (green), and catechin (black) complexes
Root-Mean-Square Fluctuation (RMSF)
The average fluctuation and variation of each amino acid in the protein target were calculated using RMSF. Figure 4 reveals that the secondary conformations of chlorogenic acid, Orlistat® and catechin remain stable during the simulation time of 100 ns. The average RMSF values for chlorogenic acid, Orlistat® complex and catechin are approximately 0.75 nm. Fluctuations were observed in residues within the range of 299-305, 403-410 in the lipase chain, and the highly fluctuated residues in the colipase are in the regions between 526-539, 547-564, and 567-587, as earlier reported by Ahmed et al., [43]. These regions of higher fluctuation belong to the secondary loop structure of the Lipase-colipase enzyme. For instance, the residues Cys237-Cys261 is a surface loop (that forms the lid domain) covering the enzyme's active sites. In conclusion, the RMSFs of all the complexes are considerably comparable to the reference protein, resulting in minimal volatility and higher stability.
Hydrogen Bond Analysis
Hydrogen bonds are particular interactions between a receptor and a ligand that are critical for the protein-ligand complex stability. In drug design, it is responsible for drug selectivity, metabolism, and adsorption [44, 45]. As seen in Figure 5, the catechin complex and chlorogenic acid complex have more hydrogen bonds throughout the simulation time. The bonds decrease from 370 to 300 and 365 to 290 in catechin and chlorogenic acids, respectively. Orlistat complex has fewer hydrogen bonds throughout the simulation period. From the H-bond analysis, we can conclude that catechin and chlorogenic acid are bound to the protein active sites effectively and tightly than Orlistat®. This indicates that the two compounds (i.e., catechin and chlorogenic acid) are better inhibitors than Orlistat®.
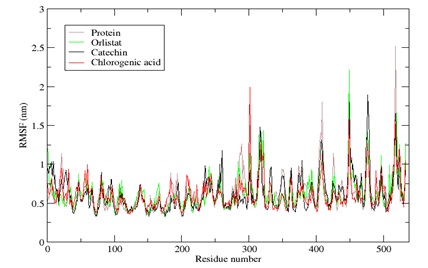 Figure 4. The backbone RMSF plot of chlorogenic acid (red), Orlistat (green) and catechin (black) complexes
Figure 4. The backbone RMSF plot of chlorogenic acid (red), Orlistat (green) and catechin (black) complexes
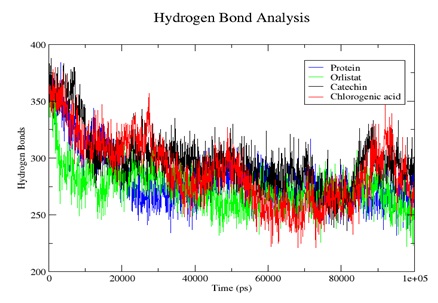 Figure 5. A plot hydrogen bond chlorogenic acid (red), Orlistat (green) and catechin (black) complexes
Figure 5. A plot hydrogen bond chlorogenic acid (red), Orlistat (green) and catechin (black) complexes
Radius of Gyration
The folding and unfolding of protein structure upon binding to the ligands can be measured by the radius of gyration (Rg). The higher the radius of gyration, the less compact (more unfolded) the protein-ligand complex. The average gyration and folding of the complexes under study is around 2.7 nm throughout the simulation. The Orlistat® complex shows some unfolding in between 30 to 40 ns and 68 to 80 ns of the simulation time. Catechin is more folded at the beginning but becomes unfolding towards the last part of the simulation. Chlorogenic acid is highly folded (Rg value generally remains constant) throughout the simulation, as shown in Figure 6. This result indicates that the catechin and chlorogenic acid complex exhibit relatively more significant behavior of compactness over the Orlistat® complex.
 Figure 6. A plot of the radius of gyration of the protein-ligand complexes. Orlistat® (green) catechin (black) and chlorogenic acid (red)
Figure 6. A plot of the radius of gyration of the protein-ligand complexes. Orlistat® (green) catechin (black) and chlorogenic acid (red)
Solvent Accessible Surface Area (SASA)
The SASA can be used to investigate interactions between the complex and the solvent throughout the 100ns MD simulation. Since the surface area significantly influences the rate of absorption and metabolism of a drug. The plot of SASA value against time for all protein-ligand complexes is shown in Figure 7. The results demonstrated that chlorogenic acid exhibited a maximum surface area at about 75 ns while catechin showed maximum surface area character at 70 ns and Orlistat at 95 ns. This is evident that catechin and chlorogenic acid will bind to the protein complex more effectively than Orlistat®.
 Figure 7. A plot of solvent accessible surface area (SASA) of the 3 ligand-protein complexes. Orlistat® (green), catechin (black), and chlorogenic acid (red)
Figure 7. A plot of solvent accessible surface area (SASA) of the 3 ligand-protein complexes. Orlistat® (green), catechin (black), and chlorogenic acid (red)
Conclusion
Sequel to the high cost and the reported side effects of Orlistat® on its short and long-term usage, it becomes imperative and challenging to develop an alternative, safer, readily available, and cost-effective drug for treating obesity. This work employed in silico techniques vis-à-vis the molecular docking, ADMET and drug-likeliness studies, and molecular dynamics methods to investigate catechin and chlorogenic acid's inhibitory tendencies and toxicity using Orlistat® as standard. The docking scores revealed a higher binding affinity of catechin and chlorogenic acid with the receptor's active site (i.e., human pancreatic lipase) compared to Orlistat®. Binding mode interactions revealed that Orlistat® has fewer contacts with the receptor's binding site, with six (6) interactions. In contrast, catechin and chlorogenic acid have (9) electrostatic interactions each.
Similarly, through the ADMET and drug-likeliness studies, it was discovered that the two compounds are non-toxic and satisfy all the criteria of a drug. Finally, molecular dynamics studies reveal the structural flexibility, stability, hydrogen bond stability, surface area, and compactness of chlorogenic acid and catechin are greater than those of Orlistat®. In conclusion, this study has validated the earlier in vivo reports of inhibitory tendencies of catechin and chlorogenic acid against human pancreatic lipase. Therefore, both are better, safer, and cheaper alternatives to Orlistat®.
Acknowledgment
The authors acknowledge the support of the Third World Academy of Science (TWAS) and International Centre for Chemical and Biological Sciences (ICCBS), Karachi, for the Postdoctoral Fellowship (FR number: 3240293180) given to S.A. Ahmed. We also appreciate the support from Ruqaiya Khalil, Urooj Quareshi, Bakhtawer Qureshi, Sajda Ashraf, and all the members of Computational Chemistry group of Prof. Dr. Zaheer Ul-Haq at PCMD, ICCBS, Karachi, Pakistan.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Sikiru Akinyeye Ahmed : 0000-0002-6402-033X
Shina Salau : 0000-0002-3067-0764
Maria Saeed : 0000-0001-6223-6375
)