Document Type : Original Research Article
Authors
Department of Pure and Industrial Chemistry, Faculty of Physical Sciences, Bayero University, Kano, Nigeria
Abstract
The compounds in this research work were studied theoretically using computational methods to analyze the inhibition of the following compounds of silicate-based obtained from Tapinanthus Globiferus. Arsenous acid, tris(trimethylsilyl)ester, Cyclotrisiloxane, hexamethyl- and Silicic acid, diethyl bis(trimethylsilyl)ester on Fe surface as Parameters were studied using quantum chemical method through DFT and molecular dynamic simulations. Mild steel Fe (111) was used due to its respective close-packed and dense atoms on the surface. The Fukui function and the local and global reactivity were calculated to give the molecule's reactivity. Based on the values of calculated adsorption and binding energies. The mechanism of the molecules was inferred to exhibit Physisorption on the Fe surface.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
Corrosion of metals is one of the adverse effects of chemistry that diverted the attention of scientists over the world [1]. The use of metals in different environments is receiving shortcomings from the harsh condition of the environment they are used. However, due to harsh conditions like high concentrations of acid, high temperature, and pressure, Metals become susceptible to corrosion; when using a hydrochloric acid solution for cleaning, pickling, or acidifying oil reservoirs. Hence, large containers of these metals are used in this sector [2-5]. Due to the production of an amphoteric protective oxide covering on its surface upon contact with the environment or aqueous solutions, aluminum is reasonably resistant to corrosion. Despite this, the higher atmosphere might still cause corrosion [3]. Most structural failures have been linked to the corrosion of metallic materials, most notably those in which mild steel is used as a supporting tool or component (alloy) [4]. Therefore, it is necessary to reduce this threat by using inhibitors to stop the metal from degrading in the environment and putting the life of that particular environment at risk [6,7].
Inhibitors compound with the hetero-atomic atoms nitrogen, sulphur, and oxygen are good inhibitors on many metals' surface. Also, some other pi-bond present in the compounds enhances the inhibition properties of the compound as the -CH- methylene of the organic compound [8].
In this research, some molecules obtained from the crude extract of Tapinanthus Globiferus Arsenous acid, tris(trimethylsilyl)ester, Cyclotrisiloxane, hexamethyl- and Silicic acid, diethyl bis(trimethylsilyl)ester were studied using DFT and molecular dynamic principles to obtained both the adsorption and binding energy of the compounds on the mild steel surface.
These molecules were selected based on the silicate present in the structures. The molecules were also selected because they have an outstanding percentage quality of about 90%, indicating that they can act on mild steel efficiently.
 Scheme 1. Showing the structures of the compounds
Scheme 1. Showing the structures of the compounds
Experimental
Quantum chemical calculations
Quantum chemistry calculations were performed using the Dmol3 modules in the Material studio 8.0 (from Accelrys Inc.) program. Dmol3 is a software that computes the electronic characteristics of molecule clusters, surfaces, and solid crystalline materials from the first principle using the density functional theory (DFT) and a numerical radial function basis set. The double numeric with polarization (DNP) basis set and the functional methods B3LYP from Becke for the exchange portion and Lee, Yang, and Parr for the correlation component were used for the density functional theory (DFT) computations. This base set was chosen because it is the best set available in Dmol3.
Per Koopman's theory, the energies of the frontier molecular orbital, the highest occupied molecular orbital (EHOMO), and the lowest unoccupied molecular orbital (ELUMO) are correlated with the ionization potential (IE) and electron affinity (EA), as shown in Equation (2) and (3) [9-17].

The dipole moment, which is equal to the magnitude of the charge (Q) at each end of the molecular dipole times the distance, is the indicator of net molecular polarity (r). Using the molecule's dipole moment (µ), it is possible to determine how many electrons are shared between two connected atoms. [18]
According to Pearson, the value of global hardness (ƞ) is roughly described as given in Equation (3). According to Equation 4, the system's global hardness (H) equals global softness (S).

The energy gap of the molecules is calculated using the relation given in Equation 5. The energy gap is a parameter that determines in the
![]()
The fraction of electrons transferred from the inhibitor to the Fe-surface, ΔN, is calculated using Equation 6. The half-electron transfer of the molecules is a parameter that demonstrates


Molecular Dynamics Simulation
To simulate a realistic portion of the surface, calculations were performed using the COMPASS FORCEFIELD and Smart ALGORITHM in a simulation box of 17 x 12 x 28 with a periodic boundary condition. The fractional depth 3.0 was used to split the Fe crystals along the (111) Plane. Before optimizing the iron surface, the lower layers' shape was limited. In order to minimize edge effects, the iron surface was then increased into a 10 x 10 supercell [1-4]. A tradeoff between a system with too much kinetic energy, where the molecule desorbs from the surface, and a system with insufficient kinetic energy, where the molecule cannot move across the surface, was made by fixing the temperature at 350 K to quench the molecules on the surface [1,2]. The NVE (microcanonical) ensemble set the temperature with a time step of 1 fs and a simulation duration of 5 ps. To obtain the statistical values of the energies on the surface of Fe, the system was programmed to quench every 250 steps (111). Different interactions were produced using forcite-tailored molecular and surface architectures. Using the relationship in Equation 14, the binding energy between the inhibitors and the Fe (111) surface was estimated in Equation 8 [19-22].
Binding Energy = Etotal − (Einhibitor + EFe surface) (8)
Results and discussion
The structure in Figure 1 represents the optimized molecule of Arsenous acid, tris(trimethylsilyl)ester, Cyclotrisiloxane, hexamethyl- and Silicic acid, diethyl bis(trimethylsilyl) ester with its density, the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital, and the optimal structure (LUMO). One technique utilized in corrosion investigations that offers crucial knowledge on the selective reactivity of the inhibitor compounds is quantum chemical calculations [10-11]. This demonstrates the possibility of interaction between the molecule and the metal surface (mild steel).
The electronic characteristics of the atoms, such as the electron density and partial charges, determine how reactive inhibitor compounds are. Electrophiles target the HOMO dense area, also serving as the active center. Whereas the LUMO orbitals depict the locations where molecules can accept electrons from the iron's d-orbital found in mild steel, Figure 2 reports the optimal structure, HOMO and LUMO orbitals, and total electron density of the molecules under investigation. The whole molecule's electron density is depicted in the picture, indicating that it can increase the adsorption capacity on metal (mild steel) surfaces [15]. The HOMO and LUMO orbital regions clearly demonstrate that the molecules' adsorption capability may depend on their HOMO and LUMO behavior. The heteroatoms found in molecules at carbonyl groups are also occupied by the HOMO and LUMO orbitals [11].
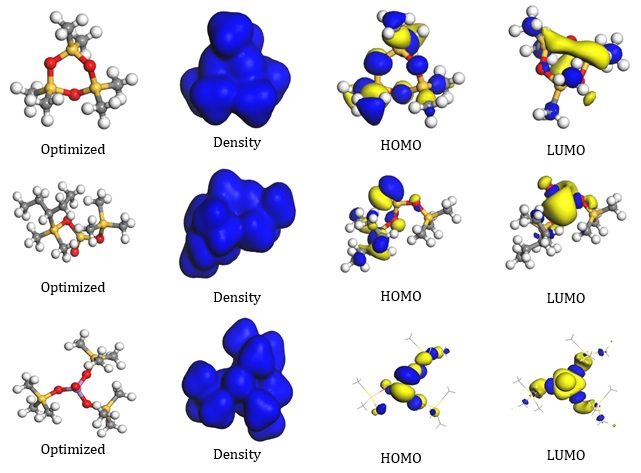
Figure 1. Showing the optimized structure, Density of the molecules, LUMO and HOMO orbitals
Table 1. Showing the Fukui functions of the molecules used for the inhibition.

From Table 1, the atoms of the molecule ATE show high potential for donating electrons to the metal surface at O2, and acceptance of electrons on the same molecule is to occur at As1. For the second compound SDB, the Si1 has more Fukui function indicating the high potential of donating electrons to the metal surface. While the atom responsible for having more potential to accept electron occurs at O2. CYE compound has its electron acceptor at Si1 and donor ability at C7. These molecules following Iorhuna et al. 2023 demonstrated that; atoms of the molecules with the highest value of the Fukui function turn to have rapid electron transfer compared to those with less value of the Fukui function [23].
Table 2 lists the molecular compounds' Outer orbital energies, together with the quantum chemical characteristics EHOMO, ELUMO, energy gap (E), dipole moment (E), electronegativity (E), global hardness, global softness, and proportion of electron transfer (N) that were examined. The ELUMO value is the lowest unoccupied molecular orbital and is typically associated with how the level at which the electrons are accepted by the molecule from the d-orbital of the metal surface. The EHOMO value is the compounds' highest occupied molecular orbital, showing a molecule's capability to denote electrons from the empty orbitals of the d-orbitals of the metal surface (mild steel). Table 2 reveals that only 3.6 electrons were transferred by the complete inhibitor molecule to the surface of the iron metal, demonstrating that the ability of these inhibitors to donate electrons to the metal surface enhanced inhibition efficiency [12,22]. Corrosion inhibition may be most effective for the molecule with the highest ΔN value.
Table 2. Showing the energy value of the molecules

Molecular Dynamics
The calculated molecular dynamic parameters explain the inhibitive potential of the molecule on the surface of the simulated metal [10,11]. In this research, the molecules were simulated on the iron to ascertain the molecule's potential on the surface. Properties such as total energy (kinetic, potential) were calculated. Surface energy, energy of the molecule, adsorption, and binding energy were calculated in Kcal/mol [12].
At the beginning of the mechanism in any corrosion inhibitor which extends the adsorption of such inhibitor is with the surface of the mild steel [10]. From the result of the molecules studied on mild steel surfaces, the negative adsorption energy on both surfaces indicates that the molecule is feasible and spontaneous on both surfaces [23]. The simulation's binding energy and adsorption energy was > -100Kcal/mol and < 100Kcal/mol, which proposed physisorption. This shows that the inhibitors are charged molecules with strong inductive and resonance effects because of the carbonyl groups and silicate present [23]. With such value, Iorhuna et al. and Eddy et al. concluded in their separate experiment that, for a mechanism to be physisorption, the value of binding energy would be less than 100 Kcal/mol [22].
Table 3. Calculated molecular dynamic simulation parameters for the studied

 Figure 2. Showing the molecule on top and side view on the surface of iron
Figure 2. Showing the molecule on top and side view on the surface of iron
Conclusions
The result and findings of the present study led to the following conclusions.
1- The molecules Arsenous acid, tris(trimethylsilyl)ester, Cyclotrisiloxane, hexamethyl- and Silicic acid, and diethyl bis(trimethylsilyl) ester are potential inhibitors for the adsorption inhibitor of mild steel.
2- The result of mild steel simulation indicated that it can undergo only the physisorption process.
That the molecules Arsenous acid, tris(trimethylsilyl)ester, Cyclotrisiloxane, hexamethyl- and Silicic acid, diethyl bis(trimethylsilyl)ester can be adsorbed to mild steel through some of its functional groups, and the electron transfer between the molecule and the surface was responsible for the inhibition which is caused by the delocalization of the pi-bond of the molecule.
Acknowledgment
The authors are grateful to the Department of Pure and Industrial Chemistry, Faculty of Physical Sciences, Bayero University Kano.
Disclosure statement
The authors declare that they have no conflict of interest
Orcid
Bishir Usman : 0000-0002-1738-4368
HOW TO CITE THIS ARTICLE
Usman Ishaq Shehu *, Bishir Usman. Corrosion Inhibition of Iron Using Silicate Base Molecules: A Computational Study, Adv. J. Chem. A, 2023, 6(4), 334-341.
DOI: 10.22034/AJCA.2023.399262.1375
)