Document Type : Original Research Article
Author
1 Biochemical Technology Program, Thamar University, Dhamar, Yemen
2 Chemistry Department, Aleppo University, Syria
Abstract
Methicillin-resistant Staphylococcus aureus (MRSA) poses an increasingly alarming threat to global public health, characterized by its escalating antibiotic resistance and heightened virulence. This dire situation necessitates the exploration of novel and hitherto uncharted targets for effective pathogen control. The primary objective of our current investigation is to identify the most promising riboswitch as a druggable target and assess its potential inhibition through both small molecules and antisense oligonucleotides (ASO) in silico. I conducted an exhaustive search for the most plausible druggable riboswitch using the RiboScan tool, subsequently subjecting its sequence to rigorous docking studies with ten distinct resveratrol derivatives through the PatchDock algorithm. In addition, we harnessed the power of ASO by designing and hybridizing five ASO sequences to silence the identified riboswitch target effectively. This comprehensive analysis pinpointed the glmS riboswitch as the most promising candidate for targeted intervention. Molecular docking results uncovered the exceptional inhibitory potential of resveratrol derivatives, with particular emphasis on ZINC000100827960, which emerged as a lead natural inhibitor. Furthermore, the ASO sequences, meticulously designed for their intended purpose, exhibited compelling efficacy in terms of hybrid free energy, RNA-RNA docking interactions, and a remarkably low probability of causing off-target effects within the human transcriptome. Taken together, our findings underscore the significant promise of both resveratrol derivatives and the ASO candidates as potent inhibitors of the glmS riboswitch in MRSA. These results warrant not only further consideration, but also rigorous experimental validation to pave the way for innovative strategies in combating the menace posed by MRSA.
Graphical Abstract
)
Keywords
- Staphylococcus aureus
- Methicillin resistant
- riboswitch
- antisense oligonucleotides
- resveratrol
- molecular docking
Main Subjects
Introduction
Staphylococcus aureus is a pathogenic bacterium that results in many organ dysfunctions, including superficial (skin) and deep ones (joints and cardiac tissues). This anaerobic, Gram-positive bacterium causes both community-acquired as well as hospital-acquired serious infections [1]. Due to the misuse of antibiotics against such highly virulent pathogen, the medical community encounters the emergence of antibiotic-resistant strains, even to the highly potent methicillin antibiotic (methicillin-resistant S. aureus, MRSA). Nowadays, more than 100 MRSA strains are classified as major of concern in USA. This led the World Health Organization (WHO) to deem MRSA as an endemic due to limited available therapeutics [2]. Vancomycin was an emergency-based therapeutic option to deal with MRSA strains but, again, vancomycin-resistant S. aureus (VRSA) was also developed and now is widely distributed in USA, India, Pakistan, and Iran [3]. It is estimated that approximately 5% of population carry MRSA. MRSA and VRSA show notorious virulence and lead to life-threatening consequences [4]. Accordingly, the search for new generation of novel targets for controlling MRSA is significantly demanding.
Bacterial genomes are classified into two main components: the core genome and the accessory genome. The core genome is composed of genes that are present in all isolates and contains essential genetic information related to cellular metabolism and replication. It makes up approximately 75% of the 2.8 Mb genome of S. aureus and is highly conserved among strains [5]. On the other hand, the accessory genome, which comprises about 25% of the total S. aureus genome, is more variable and often more strain-specific than the core genome. It consists of mobile genetic elements such as pathogenicity islands, bacteriophages, chromosomal cassettes, transposons, and plasmids acquired by horizontal transfer between strains. This is where mediators of virulence, immune evasion, and antibiotic resistance are commonly found [6,7].
One of the less-studied targets is the bacterial riboswitch. Riboswitch is a structured RNA transcribed from the 5'-untraslated region (5'-UTR) of a gene and controls gene expression from which synthesized. In other words, riboswitch is a mediator that senses the changes of some essential amino acids and metabolites in response to environmental fluctuations to increase/decrease the expression of metabolite-synthesizing gene [8].
Riboswitch offers many merits as an antibacterial target including (i) function independently of proteins or enzymes, (ii) exclusively present in prokaryotes, (iii) relatively small size (often < 100 nucleotides), and (iv) located in the 5'-UTR which conceives less impact on the coding region of the related genes [9]. With respect to structure-to-function relationship, riboswitch is composed of 2 domains: Ligand-binding domain (aptamer) and regulatory domain. Once the ligand got bound to the corresponding riboswitch in the aptamer domain, it induces conformational modifications to the overall structure facilitating, thus it is binding to the cognate gene or mRNA. This in turn blocks or triggers transcription or translation of the related gene (ON/OFF-switching) [10]. PKZ18 was demonstrated to target T-box riboswitch of MRSA with no substantial cytotoxicity toward eukaryotic cells after exposure for 24 h, but its analogues improved the specificity minimized the cytotoxicity [11]. Similarly, with an MIC (minimum inhibitory concentration) = 0.5 mg/mL, Ribocil-C was able to diminish the riboflavin uptake and its binding to flavin mononucleotide (FMN) riboswitch. This confirmed the antibacterial action versus MRSA [12].
The computational analysis of small molecules docking and antisense oligonucleotide (ASO) versus MRSA riboswitches is scarce. Hence, the present study purposes to inhibit MRSA riboswitch by small molecules and ASO.
Materials and methods
The outlined methodology used in the current theoretical study is shown in Figure 1. However, the details of every step are elucidated in detail below.
 Figure 1. Flowchart of the methodology employed in the present study
Figure 1. Flowchart of the methodology employed in the present study
Retrieval of sequence
The MRSA sequence was downloaded from the national center for biotechnology information (NCBI) [13]. The complete genome of MRSA strain M92 has an accession ID: CP015447.2 which contains ~ 3 million bp in total.
Searching for riboswitches
As an input, the full genome FASTA sequence of MRSA was uploaded to RiboScan tool (http://service.iiserkol.ac.in/~riboscan/index.html) [14] in so as to get the possible riboswitches. The tool besides finding the possible riboswitches, also ranks them by a score and detect whether the found riboswitch in the coding or complementary strands.
The tool uses a method based on profile Hidden Markov Models (pHMMs) for detecting riboswitches.
Prediction of secondary and tertiary architectures
The RiboScan tool provides also the overall secondary structure of the obtained riboswitch classes. In addition, the tertiary structure of the top-rank riboswitch was predicted from the provided sequence using 3dRNA webserver (http://biophy.hust.edu.cn/new/3dRNA/) [15]. The chosen procedure was "optimize" in order to get the best generated model. The dot-bracket pattern of the provided sequence was obtained from RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) [16].
Molecular docking
The obtained 3D structure from 3dRNA webserver was used further for molecular docking by PatchDock platform (https://bioinfo3d.cs.tau.ac.il/PatchDock/) [17]. The riboswitch structure was prepared by adding polar hydrogen and assigning charge prior to docking. This receptor was docked against 10 resveratrol derivatives in which the previous reports confirmed their potency against MRSA and its biofilm [18-20] and compared with the reference substrate, glucosamine-6-phosphate. The SDF files of the ligands were downloaded from ZINC20 dataset [21]. Only the best model, along with the native substrate, will be selected for investigation of RNA-ligands interactions by BINANA 2.0 server (https://durrantlab.pitt.edu/binana/) [22].
ADMET profiling
The absorption, distribution, metabolism, excretion, and toxicity (ADMET) of the best ligand will be predicted via admetSAR 2 web portal (http://lmmd.ecust.edu.cn/admetsar2) [23]. The server predicts Lipinski's rule of five (Ro5) as well as other toxicity parameters via inputting the SMILES of the ligand.
Design of antisense oligonucleotides (ASO)
PFRED [24] was utilized for the design of the potential ASO versus the riboswitch sequence. The program provided 199 ASOs which filtered using 4 criteria: (i) avoided 5'-untralsated region (UTR), (ii) avoided 3'-UTR, (iii) avoided polyA, and (iv) predicted ASO efficacy score > 10. The best ASO candidates will be tested for off-target with human genome/transcriptome by BLAST database (https://www.ncbi.nlm.nih.gov/geo/query/blast.html) [25]
Prediction of hybrid free energy
To evaluate the lowest hybrid free energy (HFE) upon folding the obtained ASO and their cognate strand, DuplexFold tool [26] (https://rna.urmc.rochester.edu/RNAstructureWeb/Servers/DuplexFold/DuplexFold.html) was used. However, the results were validated via RNA-RNA molecular docking tool HNADOCK (http://huanglab.phys.hust.edu.cn/hnadock/) [27].
Results
Searching for riboswitches
RiboScan output listed 9 riboswitches in the main strand and the complementary strand with a score range from 11.9 to 100.1 (Table 1). The great majority of riboswitches were found to belong to T-box class (10 riboswithces) followed by SAM1 class (3 riboswithces), Lysine, FMN, and Lysine (2 riboswitches each) whereas the remaining (glmS, Glycine, purine, and yybP-ykoY) had only one riboswitch. However, glmS exhibited the highest score (100.1) with an E-value of 6.4e-33 making it the most prominent riboswitch to be targeted in MRSA. Accordingly, only glmS riboswitch (GR) will be considered for further evaluation. The secondary architecture of the obtained all classes is illustrated in Figure 2.
Table 1. The obtained riboswitches from the provided MRSA genome sequence


Figure 2. The provided secondary structure of the main riboswitch families found in the genome of MRSA.
Molecular docking
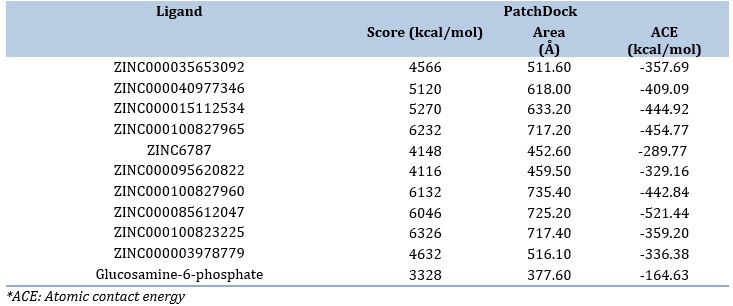
Ten resveratrol derivatives were selected as potential inhibitors to GR. PatchDock output revealed that all of the tested ligands exhibited higher binding score and atomic contact energy than the native substrate glucosamine-6-phosphate, the best of which was ZINC000100823225 with a docking score 6326 and atomic contact energy -359.20 kcal/mol (Table 2).
Table 2. PatchDock findings of resveratrol derivatives and the cogante ligand against GR
BINANA 2.0 sever unveiled the formation of 5 H-bonds with this ligand as well as 2 π–π stacking interactions. Similarly, glucosamine-6-phosphpate had only 5 H-bonds (Figure 3).
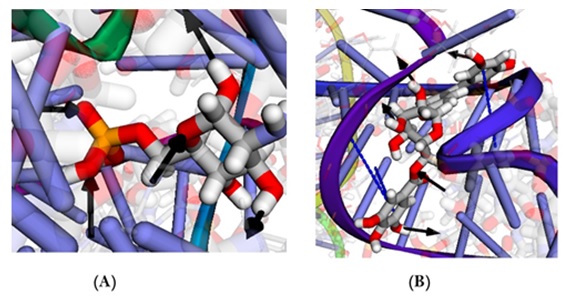 Figure 3. Cognate ligand (A) and ZINC000100823225 and (B) interaction with GR as unveiled by BINANA 2.0 server. Solid black arrow indicates hydrogen bond from donor to acceptor while dashed blue line indicates π–π stacking.
Figure 3. Cognate ligand (A) and ZINC000100823225 and (B) interaction with GR as unveiled by BINANA 2.0 server. Solid black arrow indicates hydrogen bond from donor to acceptor while dashed blue line indicates π–π stacking.
ADMET profile
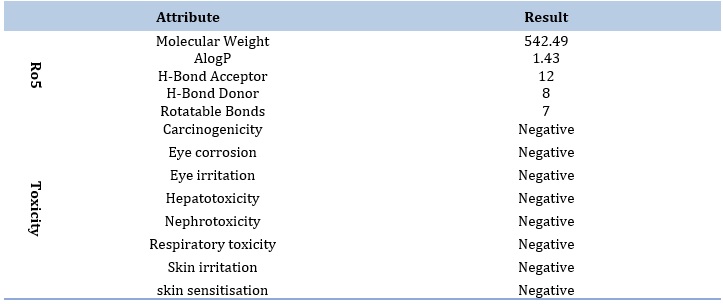
It is prominent that the best docked ligand (ZINC000100823225) was predicted to pose a good pharmacokinetic profile albeit it displayed a violation to Ro5 in terms of increased H-bond acceptor atoms (12) which was higher than the restricted level (< 10). Otherwise, the ligand is a lead candidate. In addition, the ligand also showed neither potential toxicity in any form involving carcinogenicity, hepatoxicity, nephrotoxicity, nor skin toxicity, as summarized in Table 3.
Table 3. ADMET profile of the best ligand as predicted by admetSAR 2
ASO design
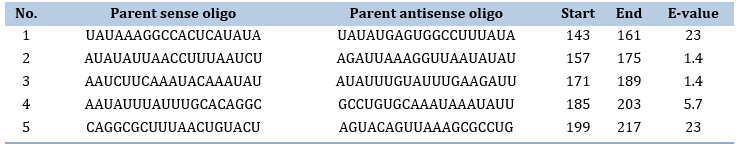
ASO design was performed using PFRED program which generated about 199 ASOs. Out of these, only 5 met the criteria of design mentioned in the methodology section. The ASOs are presented in Table 4. Next, the top 5 ASO were checked for off-target with human transcriptome. Fortunately, all of them were above the set threshold (E-value < 0.5). This confirms the good design and selection of the program.
Table 4. Designed ASO along with Off-target E-value
HFE prediction
As demonstrated in Figure 4, ASO5 followed by ASO4 were the best in terms of complementarity strength to GR parent sense sequence giving HFE of -26.5 and -32.4 kcal/mol. However, ASO2 and ASO3 showed weak complementarity strength (-20.6 and -21.1) (Figure 4). This made ASO5 as the best potential ASO to silence GR. To validate those findings, RNA-RNA docking was made via HNADOCK platform.
 Figure 4. Sense-Antisense complementation and the predicted HFE.
Figure 4. Sense-Antisense complementation and the predicted HFE.
The docking scores were inconsistent with DuplexFold output, ranking ASO1 as the best while ASO5 was the lowest (-365.68 vs. -218.64 kcal/mol). ASO2 and ASO3 were in-between (Figure 5). The difference in findings could be attributed to the fact that HFE is calculated based on sequence-based, bases hybridization whilst RNA-RNA docking is predicted according to structure-based complementation. In general, this leads us to the fact that all ASO are significantly potential GR silencers in silico, but the in vitro assays will give the most crucial result.
 Figure 5. RNA-RNA docking along with the docking score of each ASO-GR complex.
Figure 5. RNA-RNA docking along with the docking score of each ASO-GR complex.
Discussion
MRSA is a predator bacterial pathogen responsible for serious health issues and death cases each year particularly in USA and Europe. It still represents a medical obstacle since its discovery more than 60 years ago [28]. In fact, MRSA had emerged even before the introduction of methicillin into pharma market [29]. Riboswitches sense the concentration of a significant intracellular metabolite needed by an indispensable metabolic pathway in response to the environmental variation. Therefore, targeting bacterial riboswitches represent a new era for the development and testing novel drug candidate either of natural or synthesizable origins [30]. This instigates the medical community to search for novel target and novel drugs to deal with such threat. Recent attention was directed toward MRSA riboswitch as a novel druggable target. Wang et al. [12] targeted FMN riboswitch with small molecules and proved their efficacy as potential drugs. Likewise, Traykovska and Penchovsky [31] designed and successfully applied ASO to silence S-adenosyl methionine-1 riboswitch of MRSA as a novel therapy. Theoretically, numerous studies reported the exploitation of bioinformatics approaches to provide unprecedented targets as well as inhibitors to the emerging pathogens including MRSA. This includes chalcone derivatives [32], benzamide derivatives [33], diverse inorganic coordination complexes [34], 2-Arylbenzimidazole Derivatives [35], and some natural flavonoids [36]. In this study, GR was the target by small molecules (resveratrol derivatives) as well as ASO to inhibit the formed GR and prevent the transcription of GR. A dozen of resveratrol was docked into GR and proved their potency as potential natural inhibitors of such target. Besides, 5 ASO were designed against GR sequence and demonstrated their efficiency using HFE predicted by DuplexFold and docking to the native GR 3D structure by HNADOCK. GR serves as regulator of glucosamine synthase gene expression one of the contributing players to peptidoglycan biosynthesis. Upon binding its cognate ligand glucosmine-6-phosphate, a conformational change ensues leading to its activation and thus transformation to self-cleaving ribozyme. Ultimately, GR ribozyme activity cleaves glucosamine synthase mRNA in preparation to its decay by RNase J1 [37,38]. Recently, Mujwar and Pardasani [39] utilized computational methods to identify and virtually screen drug library against Mycobacterium tuberculosis riboswitches. Similarly, Anthrax riboswitches were also targeted as a means to inhibit and eradicate Bacillus anthracis bacterium in silico [40]. Furthermore, Salmonella enterica was another bacterium to be targeted via its riboswitch via a set of drug library in silico [41]. So, targeting such important genetic target to prevent MRSA membrane biosynthesis is an unprecedented aspect and the current study output deserves further validation in vitro.
Conclusion
The present theoretical study elucidates the inhibition of Methicillin-resistant Staphylococcus aureus (MRSA) growth by a series of resveratrol derivatives, among which ZINC000100823225 emerges as the most promising potential inhibitor. This lead candidate exhibits a notable characteristic of adhering to of Lipinski's Rule of Five (Ro5) and demonstrates an absence of potential toxicity. Furthermore, I have designed and computationally demonstrated the efficacy of five Antisense Oligonucleotides (ASO), which exhibit a significant capacity to silence the previously unexplored target, GR, in silico. This silencing potential is underscored by high hybridization energy and docking scores, as predicted by HNADOCK, while simultaneously ensuring the absence of any plausible off-target effects, particularly with respect to human genes. The combined utilization of these two strategic approaches holds substantial promise for mitigating the prevalence of MRSA and, as such, warrants further rigorous laboratory experimentation.
List of abbreviations
ADMET Absorption, distribution, metabolism, excretion, and toxicity
ASO Antisene oligonucletoides
FMN Flavin mononucleotide
GR glms riboswitch
HFE Hybrid free energy
MRSA Methicillin-resistant staphylococcus aureus
VRSA Vancomycin-resistant S. aureus
WHO World Health Organization
Ethics Approval and Consent to Participate
Not applicable.
Human and Animal Rights
Not applicable.
Conflict of interest
The author declares no known conflicts of interest.
Disclosure statement
The authors declare that they have no conflict of interest.
Orcid
Haitham Ahmed Al-Madhagi : 0000-0002-3850-244X
HOW TO CITE THIS ARTICLE
Haitham Ahmed Al-Madhagi*. Empowering Therapeutic Strategies against Methicillin-Resistant Staphylococcus Aureus Riboswitch: Unveiling the Potential of Small Molecules and Antisense Oligonucleotides through In Silico Analysis. Adv. J. Chem. A, 2024, 7(1), 15-26.
)